
| Synonym: | Fmoc-L-Photo-Leucine |
| CAS #: | 1360651-24-6 |
| Molecular Formula: | C20H19N3O4 |
| Molecular Weight: | 365.4 |
| Fmoc-L-Photo-Leucine (CAS #: 1360651-24-6) is a Fmoc-protected, diazirine-containing photoreactive analogue of the canonical amino acid L-leucine. The compound integrates a 3-methyl-3H-diazirine ring into the side chain of leucine, generating a highly reactive singlet carbene species upon irradiation with near-UV light (~345–365 nm). This carbene inserts non-selectively into C–H, N–H, O–H, and C–C bonds in neighboring molecules, forming stable covalent adducts that covalently capture transient, non-covalent molecular interactions. The Fmoc protecting group on the α-amino function renders Fmoc-L-Photo-Leucine fully compatible with standard Fmoc solid-phase peptide synthesis (SPPS), enabling its site-specific incorporation into peptides, peptidomimetics, and photoaffinity probe molecules.Originally described by Suchanek, Radzikowska, and Thiele (Nature Methods, 2005), photo-leucine has since become an indispensable tool in chemical biology for the identification of protein–protein interactions (PPIs), drug target identification, structural proteomics, and chemoproteomics. The Fmoc-protected form critically extends the utility of this chemistry beyond metabolic cell labeling to rationally designed, sequence-defined photoaffinity probes and peptidomimetic drugs. This review comprehensively surveys the chemical identity, structural features, photochemical mechanism, synthetic routes, analytical characterization methods, incorporation strategies, documented research applications, comparative advantages and limitations, handling requirements, and future prospects of Fmoc-L-Photo-Leucine, drawing on the primary literature and commercial product documentation. 1. Introduction and Scientific Context 1.1 The Challenge of Capturing Transient Protein Interactions Protein–protein interactions (PPIs) and the interactions between small-molecule probes or drugs and their biological targets underpin virtually every cellular signaling pathway, metabolic process, and disease mechanism. Yet the biochemical toolkit available to characterize such interactions in their native cellular environment has historically been constrained by a fundamental limitation: most biochemical methods detect interactions only after cell lysis, disrupting the delicate balance of concentrations, post-translational modifications, and compartmentalization that governs interaction specificity in living systems. Affinity pull-down assays, co-immunoprecipitation, two-hybrid screening, and proximity ligation assays all suffer to varying degrees from false positives arising from post-lysis reassortment of proteins and false negatives owing to the loss of transient complexes during sample preparation. Photo-crosslinking chemistry addresses these limitations by providing a means to lock transient, non-covalent interactions into stable, covalent bonds in living cells or in native biochemical environments, prior to any perturbation of the system. A photoactivatable probe is introduced into the cellular milieu, allowed to engage its interaction partner through normal non-covalent forces, and then converted to a covalent crosslinker by a brief pulse of UV light. The resulting covalent complex is stable to denaturing conditions, enabling rigorous downstream analysis by SDS-PAGE, western blotting, or mass spectrometry-based proteomics. 1.2 Historical Development of Photo-Leucine The concept of photoaffinity labeling dates to the 1962 work of Singh and Westheimer, who demonstrated that a diazo compound could label the active site of chymotrypsin upon photolysis. Over subsequent decades, the field evolved through successive generations of photoreactive functional groups—aryl azides, benzophenones, and, more recently, diazirines—each offering a distinct profile of reactivity, stability, cell compatibility, and structural perturbation. The specific use of photo-reactive amino acid analogues—molecules that can be incorporated directly into proteins by the cellular translational machinery—was first demonstrated convincingly by Suchanek, Radzikowska, and Thiele at the Max Planck Institute in Dresden, published in Nature Methods in 2005. They designed two new photoactivatable amino acids, photo-methionine and photo-leucine, whose structures closely mimic those of the corresponding natural amino acids. This structural mimicry is critical: it allows the photo-amino acids to evade the stringent quality-control mechanisms of aminoacyl-tRNA synthetases and be incorporated into proteins by the unmodified mammalian translation machinery, achieving global labeling of the cellular proteome. Upon UV irradiation of metabolically labeled cells, multiple known and previously unknown protein–protein interactions—including a novel interaction between the progesterone membrane receptor PGRMC1 and the cholesterol-sensing protein Insig-1—were detected. The Fmoc-protected derivative of L-photo-leucine constitutes a second-generation tool, extending the utility of photo-leucine chemistry from metabolic whole-cell labeling to rational, site-specific probe design via Fmoc-SPPS. By enabling the precise placement of a single photo-leucine residue at any position in a synthetic peptide, Fmoc-L-Photo-Leucine provides the researcher with directional, hypothesis-driven control over where crosslinking occurs, dramatically simplifying data interpretation and enabling highly specific photoaffinity labeling experiments. 2. Chemical Identity and Physicochemical Profile 2.1 Nomenclature and Registry Information IUPAC Name: (2S)-2-(9H-fluoren-9-ylmethoxycarbonylamino)-3-(3-methyldiazirin-3-yl)propanoic acid CAS Number: 1360651-24-6 MDL Number: MFCD31380715 Molecular Formula: C20H19N3O4 Molecular Weight: 365.4 g/mol Synonyms: Fmoc-Photo-Leu; Fmoc-Photo-Leu-OH; L-PhotoLeu-OH(Fmoc); Fmoc-L-photo-leucine Stereochemistry: (S)-configuration UV Absorption (λmax): ~265–270 nm (Fmoc) and ~345 nm (diazirine) Photoactivation Wavelength: ~345–365 nm (UV-A) Solubility: DMF, DMSO, DCM (well soluble); limited aqueous solubility Storage Conditions: -20°C, desiccated, protected from light (amber vial) 2.2 Structural Architecture: Three Functional Domains The molecule consists of three structurally and functionally distinct regions, each contributing essential properties: 2.2.1 The L-Amino Acid Backbone The central α-amino acid scaffold is a two-carbon chain bearing: (1) a free carboxyl group (–COOH) available for peptide bond formation during SPPS; (2) an α-amino function protected by the Fmoc group; and (3) the (S)-absolute configuration at the α-carbon, matching the stereochemistry of natural L-leucine. This L-configuration is essential: only the L-form is recognized with high fidelity by aminoacyl-tRNA synthetases for metabolic incorporation, and L-amino acid residues are the universal building blocks for bioactive peptides. Racemization at the α-carbon during synthesis must be minimized, as D-photo-leucine is unlikely to be recognized by cellular machinery and its presence would dilute the effective concentration of the photoactivatable probe. 2.2.2 The 3-Methyl-3H-Diazirine Photophore The defining structural and functional element of Fmoc-L-Photo-Leucine is the 3-methyl-3H-diazirine ring. Chemically, the diazirine ring is a three-membered cyclic system comprising one carbon atom flanked by two nitrogen atoms; the two nitrogens are connected to each other through an internal N=N double bond while each is also connected to the central carbon through a single C–N bond. In the 3-methyl-3H-diazirine subtype present in photo-leucine, the carbon of the diazirine ring bears one methyl substituent; the ring is attached to the leucine backbone via a methylene (–CH2–) tether. This geometry positions the diazirine ring at approximately the same spatial location as the branching isobutyl side chain of leucine—contributing to the leucine-isostere character of the compound. The methyl group on the diazirine carbon mimics one of the two terminal methyl groups of leucine, preserving hydrophobic character and approximate molecular volume. The diazirine ring itself is the smallest known photoreactive organic group, with a molecular mass contribution of only ~40 Da above the leucine side chain. This minimality is crucial: it minimizes steric and electronic perturbation of peptide and protein structures, maximizing the probability that photo-leucine-substituted molecules retain the binding affinity and specificity of the parental leucine-containing sequence. 2.2.3 The Fmoc Protecting Group The 9-fluorenylmethyloxycarbonyl (Fmoc) group is the industry-standard base-labile protecting group for the α-amine during Fmoc-SPPS. It is introduced onto the α-amino group as a carbamate. During SPPS, Fmoc removal is accomplished by treatment with 20% piperidine in DMF (or other secondary amines), which proceeds via a β-elimination/cyclization mechanism to release dibenzofulvene and CO₂, exposing the free amine for the next coupling cycle. The Fmoc group is orthogonal to the acid-labile side-chain protecting groups used in Fmoc-SPPS strategy, allowing selective Fmoc deprotection without disturbing other protecting groups or the diazirine ring. 2.3 Comparison to L-Leucine L-Leucine differs from photo-leucine exclusively in the side chain: leucine carries a 2-methylpropyl (isobutyl) group (–CH2–CH(CH3)2), while photo-leucine carries a –CH2–C(CH3)(N=N) diazirine group. The key structural analogy is that both side chains are aliphatic and nonpolar; both present a methyl-bearing carbon approximately 3 bonds from the β-carbon; and both have comparable van der Waals volumes. This near-identity of steric and electronic properties underpins the ability of photo-leucine to be metabolically incorporated into proteins by the mammalian translation machinery, evading recognition-based exclusion by leucyl-tRNA synthetase. 3. Photochemical Mechanism and Reactivity 3.1 UV Absorption and Photoexcitation Fmoc-L-Photo-Leucine exhibits two UV absorption regions. The Fmoc chromophore absorbs at ~265–270 nm (π→π*, ε ~ 5,000–6,000 M⁻¹cm⁻¹) and at ~301 nm (a weaker secondary band); these bands disappear upon Fmoc removal. The diazirine chromophore absorbs at approximately 340–350 nm (n→π*, ε ~ 100–200 M-1cm-1)—a weak but diagnostically useful band attributable to the N=N system of the diazirine ring. This absorption in the UV-A range is well separated from typical protein and nucleic acid chromophores (aromatic amino acids at ~280 nm; nucleic acids at ~260 nm), minimizing non-specific UV-induced damage to biological molecules during photocrosslinking experiments. Irradiation at 345–365 nm—typically delivered by UV-A lamps, LED arrays, or filtered mercury-vapor light sources—selectively excites the diazirine chromophore, triggering photolysis without significantly exciting aromatic amino acids or nucleotides. 3.2 Photolysis and Carbene Generation: The Two-Step Mechanism Recent mechanistic work (Nature Communications, 2024) has clarified that the photolysis of alkyl diazirines such as photo-leucine proceeds through a two-step sequential pathway rather than a direct concerted process. Upon photoexcitation of the diazirine ring: • Step 1 — Diazo intermediate: The diazirine ring opens to form a diazo compound (R–C(CH₃)=N₂), a metastable species that absorbs at shorter wavelengths (~340 nm) and is longer-lived than the subsequent carbene. The diazo intermediate is preferentially reactive toward polar residues, especially acidic residues (Asp, Glu) and buried polar side chains, through a relatively selective electrophilic mechanism. • Step 2 — Free carbene: Further photolysis (or thermal decomposition) of the diazo compound liberates molecular nitrogen (N₂) and generates a free alkyl carbene—a carbon species bearing only six electrons in two orbitals (a filled σ-type orbital and an empty p-type orbital). The singlet carbene is the primary reactive intermediate for non-selective crosslinking. It has a half-life on the nanosecond timescale and reacts with essentially any chemical bond in its immediate vicinity through electrophilic insertion reactions. This mechanistic insight has significant practical implications: the relative contribution of the diazo pathway versus the carbene pathway can be tuned by adjusting irradiation wavelength and light intensity. Irradiation at longer wavelengths and lower intensities favors diazo-mediated crosslinking, providing higher selectivity for polar residues. Irradiation at shorter wavelengths or higher intensities drives complete conversion to the carbene, providing the non-selective, broad-spectrum crosslinking for which photo-leucine is most widely used. 3.3 Carbene Insertion Reactions The singlet carbene generated from photo-leucine reacts through concerted, pericyclic insertion into a wide range of chemical bonds: • C–H bond insertion: The most common pathway in protein environments. The carbene inserts into aliphatic and aromatic C–H bonds, forming a new C–C bond. This non-selective reactivity is critical for capturing hydrophobic interfaces and transmembrane contacts where nucleophilic residues are absent. • N–H bond insertion: Insertion into N–H bonds of amide backbones, lysine side chains, or arginine guanidinium groups, forming stable C–N bonds. • O–H bond insertion: Reaction with serine, threonine, and tyrosine hydroxyl groups, water molecules, or ester oxygens, forming C–O bonds. • C=O and C–C bond insertion: Less common but documented, particularly relevant for insertion into peptide bonds or lipid acyl chains. Because the carbene lifetime is on the order of nanoseconds, only molecules within a contact distance of a few Ångströms at the moment of photolysis are captured. This temporal and spatial precision provides the near-zero-length crosslinking characteristic of photo-leucine, delivering structural information about direct intermolecular contacts rather than proximity within a larger diffusion radius. 3.4 Competing Reactions and Quenching Several competing pathways reduce overall crosslinking yield. In aqueous environments, the carbene is rapidly quenched by water through O–H insertion, producing a gem-diol or alcohol product rather than a protein crosslink. Intersystem crossing from the singlet to the triplet carbene state results in a more stable but less reactive species that preferentially abstracts hydrogen atoms rather than inserting into bonds. Intramolecular rearrangements of the carbene can also occur, reducing the effective concentration of reactive carbene. These competing pathways typically limit gross crosslinking yields to 10–40% under optimized conditions, and productive crosslinking with a defined binding partner may be still lower depending on interaction affinity and geometry. Key Insight: Performing photocrosslinking on ice (0–4°C) significantly improves yields by slowing water diffusion and aqueous quenching of the carbene intermediate, extending the effective half-life of productive crosslinking chemistry. 3.5 Stability Under Chemical Synthesis Conditions A critical practical consideration is the stability of the diazirine ring under the conditions of Fmoc-SPPS. Extensive experimental evidence confirms that the diazirine ring of Fmoc-L-Photo-Leucine is stable under all standard Fmoc-SPPS conditions: • Fmoc deprotection: 20% piperidine in DMF (1 × 5 min, 1 × 10 min) — stable. • Coupling reactions: HATU/DIPEA, HBTU/DIPEA, PyBOP/NMM at room temperature — stable. • Acid-mediated global deprotection and cleavage: TFA/TIS/H2O (95:2.5:2.5, 2–4 h at RT) — stable. • HPLC purification: Aqueous acetonitrile/0.1% TFA or 0.1% formic acid gradients — stable. • Lyophilization and storage in DMF or DMSO solutions — stable (dark, cold). 4. Synthesis and Preparation 4.1 Historical Synthetic Routes The synthesis of L-photo-leucine has evolved significantly since its introduction. The original route (Suchanek et al., 2005) started from 4,4′-azipentanoic acid, proceeding through α-bromination of the azi-carboxylic acid by treatment with N-bromosuccinimide, followed by nucleophilic aminolysis of the α-bromo acid in ammonia-saturated methanol. This route furnished racemic (D,L)-photo-leucine; the desired L-enantiomer was obtained by enzymatic resolution of the racemic mixture. The overall yield was low and the route required several chromatographic purifications, limiting its practical utility for preparative-scale synthesis. MacKinnon, Garrison, Hegde, and Taunton (JACS, 2007) reported an improved synthesis of photo-leucine that offered better overall yield and eliminated the need for enzymatic resolution. Their approach employed ozonolysis of a commercially available leucine derivative to introduce the ketone function required for diazirine ring formation, followed by installation of the diazirine through the ammonia/hydroxylamine-O-sulfonic acid (HOSA) method developed by Church and Weiss. This route supposes a significant improvement over the original six-step synthesis, which proceeded in low yield and required enzymatic resolution of a racemic intermediate. 4.2 Modern Stereoselective Synthesis Current best practice for the synthesis of enantiomerically pure Fmoc-L-Photo-Leucine generally employs one of two strategic approaches: • Chiral pool strategy: Starting from commercially available L-amino acid derivatives bearing a β-keto function or ozonizable side-chain olefin, the diazirine is installed after establishing the stereocentre. This approach avoids enzymatic resolution and produces the L-enantiomer with high e.e. • Asymmetric synthesis: Chiral auxiliary-controlled alkylation or asymmetric phase-transfer catalysis is used to install the (S)-configured α-amino function with high enantioselectivity, after which the diazirine ring is constructed on the side chain using the HOSA oxidation–cyclization sequence. The diazirine ring formation via the HOSA method involves: (i) reaction of a ketone precursor with ammonia to form an imine; (ii) cyclization under oxidizing conditions using hydroxylamine-O-sulfonic acid to form the diaziridine ring (a 3-membered C–N–N ring); and (iii) base-mediated oxidation (e.g., with triethylamine and iodine) to introduce the N=N double bond, completing the diazirine ring. The entire sequence proceeds under mild conditions compatible with amino acid chemistry. Once enantiomerically pure H-L-Photo-Leucine (the free amino acid) is obtained, introduction of the Fmoc group is straightforward. Treatment of the free amino acid with Fmoc-OSu (Fmoc-N-hydroxysuccinimide ester) in aqueous sodium bicarbonate/dioxane or acetone affords Fmoc-L-Photo-Leucine in good yield and high purity after recrystallization or column chromatography. 5. Analytical Characterization 5.1 UV-Visible Spectroscopy The most diagnostically useful spectroscopic feature of Fmoc-L-Photo-Leucine is the diazirine n→π* absorption band at approximately 340–350 nm. This band, although weak (ε ~ 100–200 M⁻¹cm⁻¹), is highly characteristic and can be used to: confirm diazirine integrity in freshly prepared solutions; monitor premature photolysis during synthesis or handling; quantify diazirine-containing peptides in HPLC fractions by UV detection at 345 nm; and optimize photocrosslinking conditions by monitoring the time-course of absorbance decrease at 345 nm during UV irradiation. Upon Fmoc removal, the absorption at 265–301 nm disappears, while the diazirine band at 345 nm persists in the deprotected peptide. This spectral change provides a simple check that Fmoc deprotection was complete while confirming that the photophore survived the deprotection step. 5.2 Nuclear Magnetic Resonance Spectroscopy ¹H NMR spectroscopy of Fmoc-L-Photo-Leucine in DMSO-d6 is characterized by the following resonances: • Fmoc aromatic protons: complex multiplets at δ 7.30–7.90 ppm (8H, aryl H) • Fmoc methine (9-CH): triplet at δ ~4.22 ppm (1H, CH of fluorene) • Fmoc methylene (OCH₂): doublet of doublets at δ ~4.15–4.35 ppm (2H, OCH₂) • α-CH: multiplet at δ ~3.95–4.05 ppm (1H, overlapping with Fmoc CH₂) • β-CH₂: AB quartet or multiplet at δ ~1.90–2.10 ppm (2H, diastereotopic CH₂) • Diazirine methyl (C-CH₃): singlet at δ ~1.25–1.35 ppm (3H, characteristic diazirine methyl) • NH: broad doublet at δ ~7.55–7.80 ppm (1H, exchangeable with D₂O) 13C NMR shows the characteristic quaternary diazirine carbon at δ ~25–28 ppm and the diazirine methyl carbon at δ ~18–20 ppm. The α-carbon resonance appears at δ ~51–53 ppm, and the carboxyl carbon at δ ~172–174 ppm. The Fmoc carbonyl (carbamate C=O) appears at δ ~155–157 ppm. 5.3 Mass Spectrometry ESI-MS (positive mode) of Fmoc-L-Photo-Leucine reveals characteristic ions: [M+H]⁺ at m/z 366.14, [M+Na]⁺ at m/z 388.12, and [M+K]⁺ at m/z 404.09. High-resolution mass spectrometry (HRMS) confirms the molecular formula C20H19N3O4 with sub-ppm mass accuracy. MALDI-TOF analysis with α-cyano-4-hydroxycinnamic acid (CHCA) matrix shows the [M+H]⁺ and [M+Na]⁺ ions. When Fmoc-L-Photo-Leucine is incorporated into peptides, mass spectrometry of the peptide confirms incorporation through the expected mass addition of 365.14 Da per residue (replacing leucine at 131.09 Da, net mass addition +234.05 Da). LC-MS/MS fragmentation of photo-leucine-containing peptides yields characteristic b- and y-ion series that can be used to confirm the position of incorporation within the peptide sequence. 5.4 Chiral HPLC and Optical Rotation The stereochemical integrity of the (S)-configuration is confirmed by chiral HPLC analysis (typically using a Chiralpak column) and optical rotation measurement. Commercial lots of Fmoc-L-Photo-Leucine are specified at ≥98% enantiomeric excess (e.e.) by chiral HPLC. The optical rotation of photo-leucine derivatives is negative (levorotatory), consistent with L-configuration. Certificates of Analysis from major suppliers typically include optical rotation data to confirm stereo purity. 6. Incorporation Strategies 6.1 Fmoc Solid-Phase Peptide Synthesis (SPPS) The primary designed application of Fmoc-L-Photo-Leucine is as a building block for Fmoc-SPPS. In this approach, the compound is used exactly like any other Fmoc-amino acid building block. Dissolved in DMF (typically at 0.2–0.5 M concentration), it is activated using standard coupling reagents (HATU, HBTU, PyBOP, or COMU) in the presence of a base (DIPEA or NMM, 2–4 equivalents relative to the amino acid), and coupled to the free N-terminus of the growing resin-bound peptide chain. Double coupling is sometimes employed for difficult sequences or for sterically demanding positions. The ability to incorporate photo-leucine at any desired position in a synthetic peptide sequence is the key advantage of the Fmoc-SPPS approach over metabolic global labeling. By positioning the photo-leucine residue at the site expected to be closest to the binding partner—guided by structural data, modeling, or alanine scanning—the researcher generates a directional, sequence-defined crosslinker. This site-specific approach dramatically simplifies crosslink identification by mass spectrometry, since only a single photoreactive site is present in the probe. Importantly, the diazirine ring is fully stable to all Fmoc-SPPS conditions: Fmoc deprotection, coupling, side-chain deprotection, and TFA-mediated resin cleavage. Peptides are purified under standard RP-HPLC conditions with UV detection, taking care to limit UV exposure to routine analytical wavelengths (214 nm or 254 nm) and to handle purified fractions under amber lighting or in the dark. 6.2 Metabolic Incorporation in Living Cells The unprotected free amino acid form of L-photo-leucine can be incorporated globally into the cellular proteome through metabolic labeling. This strategy exploits the substrate permissiveness of leucyl-tRNA synthetase (LeuRS), which tolerates the photo-leucine side chain sufficiently to mischarg tRNA^Leu, allowing ribosomal incorporation of photo-leucine in place of leucine during ongoing protein synthesis. The standard protocol involves growing cells in leucine-deficient (and often isoleucine- and valine-deficient) medium supplemented with photo-leucine (4 mM) and photo-methionine (1.7 mM) for approximately 22 hours. This dual-labeling approach maximizes the probability that any given protein will have incorporated at least one photoreactive residue, since leucine and methionine together account for ~11% of average protein sequence composition. Following labeling and washing, cells are UV-irradiated (310–370 nm, 1–10 minutes on ice) and lysed for analysis. The Fmoc-protected form is specifically designed for chemical synthesis, not metabolic incorporation; however, understanding both routes is important for experimental design. When metabolic incorporation provides sufficient labeling density for the biological question, the simpler metabolic approach is preferred. When site-specific probe design is required—for drug target identification, epitope mapping, or the study of specific PPIs—the Fmoc-SPPS route using Fmoc-L-Photo-Leucine provides superior precision. 6.3 High-Yield Expression in E. coli For structural proteomics and cross-linking mass spectrometry applications requiring milligram quantities of photo-leucine-labeled proteins, Iacobucci et al. (Journal of Proteome Research, 2020) developed an optimized protocol for recombinant expression of photo-leucine-labeled proteins in Escherichia coli. By growing cells to high density before initiating amino acid depletion and photo-amino acid supplementation, they achieved up to 34% incorporation of photo-leucine (replacing leucine) into proteins of interest, obtaining up to 3 mg of labeled protein per 100 mL culture—a tenfold improvement in efficiency over standard protocols. The method was demonstrated for the chaperones trigger factor and SecB, and the resulting photo-leucine-labeled proteins yielded 12 UV-crosslinks for trigger factor that were fully consistent with the published crystal structure. 6.4 Semi-Synthetic Protein Preparation by Native Chemical Ligation For large proteins where total chemical synthesis is impractical, semi-synthetic approaches combining recombinant protein expression with chemical peptide synthesis can be used to incorporate Fmoc-L-Photo-Leucine at defined sites. In native chemical ligation (NCL), a synthetic peptide thioester containing the photo-leucine residue is ligated to a recombinant protein fragment bearing an N-terminal cysteine, forming a native amide bond at the ligation junction. Expressed protein ligation (EPL) extends this to biosynthetically generated thioesters via inteins. Vila-Perelló et al. demonstrated the power of this approach for photo-methionine incorporation into the Smad2 signaling domain, enabling covalent capture of phosphorylation-dependent protein oligomerization events that could not be captured by conventional methods. 6.5 Incorporation via Genetic Code Expansion Genetic code expansion (GCE) using orthogonal aminoacyl-tRNA synthetase/tRNA pairs in principle allows site-specific, in vivo incorporation of photo-leucine at amber stop codons (UAG). While GCE has been successfully applied to other diazirine-containing amino acids (e.g., photo-lysine, and aryl-diazirine amino acids), metabolic incorporation by native LeuRS currently remains the most practical approach for photo-leucine in mammalian cells, as the substrate permissiveness of native LeuRS provides global labeling without requiring synthetase engineering. 7. Research Applications 7.1 Identification of Protein–Protein Interactions in Living Cells The foundational application of photo-leucine, as demonstrated by Suchanek et al. (2005), is the covalent capture and identification of protein–protein interactions (PPIs) in living cells or intact cellular environments. By metabolically incorporating photo-leucine and photo-methionine into the proteome of COS7 monkey kidney cells, the authors UV-irradiated intact cells and demonstrated that crosslinked protein complexes could be detected by western blotting with a sensitivity and specificity superior to conventional pull-down approaches. Critically, the technique revealed a novel direct interaction between the progesterone membrane receptor PGRMC1 and Insig-1, a protein involved in sterol-mediated regulation of HMGCR—an interaction with direct relevance to lipid metabolism and cholesterol homeostasis. Since the original publication, photo-leucine-based PPI identification has been applied across a wide range of biological systems, including investigations of membrane protein complexes, ribosome-associated proteins, chaperone networks, and signal transduction assemblies. The key advantage over biochemical pull-downs is that interactions are locked in covalently before cell lysis, preventing artifactual complex formation or dissociation. 7.2 Drug Target Identification and Validation The identification of the molecular target(s) of bioactive small molecules—both natural products and synthetic drug candidates—is a critical and often rate-limiting step in drug discovery. Photoaffinity labeling with photo-leucine-containing probes has emerged as one of the most powerful approaches to this challenge. MacKinnon, Garrison, Hegde, and Taunton (JACS, 2007) provided a landmark demonstration of this strategy. They incorporated photo-leucine into the cyclodepsipeptide inhibitor of cotranslational translocation (a natural product that blocks protein translocation into the ER), at a position that was determined by structure–activity relationship analysis to be tolerated by the target. Photoaffinity labeling in a crude microsomal membrane fraction, followed by copper-catalyzed azide–alkyne cycloaddition (CuAAC) “click chemistry” with rhodamine–azide reporter tag (a bifunctional probe approach using the alkyne handle in the probe), identified Sec61α—the pore-forming subunit of the Sec61 translocon—as the molecular target. This target identification was achieved against the background of a complex proteome in membrane fractions, underscoring the selectivity and power of the photo-leucine approach. Similarly, van der Meijden and Robinson (Journal of Peptide Science, 2015) synthesized a photo-leucine-containing derivative of polymyxin—a last-resort antibiotic against Gram-negative bacteria—and used it in photolabeling studies to probe the molecular targets of polymyxin in Escherichia coli. The study illuminated the mechanism of action of polymyxins, which is important for understanding and potentially circumventing polymyxin resistance. 7.3 GPCR and Membrane Protein Interaction Studies G protein-coupled receptors (GPCRs) are the largest family of drug targets in the human genome, yet the precise molecular contacts through which peptide ligands, lipopeptide pepducins, and small molecules engage GPCRs have been challenging to map by conventional means, partly because the interactions often involve transmembrane or intracellular hydrophobic surfaces not amenable to amine-reactive crosslinking. Janz, Ren, Looby, Kazmi et al. (JACS, 2011) used photo-leucine as a key element to directly confirm the interaction of a pepducin—a cell-penetrating lipopeptide GPCR modulator—with its target receptor CXCR4. They synthesized an analogue of the CXCR4 pepducin agonist ATI-2341 in which the Leu residue at position 7 was replaced with L-photo-leucine (incorporated by Fmoc-SPPS). Structure–activity studies confirmed that this substitution was tolerated without loss of receptor agonism. UV irradiation of cells expressing CXCR4-GFP fusion proteins in the presence of the photo-leucine pepducin produced a specific, UV-light-dependent crosslinked band corresponding to a pepducin-CXCR4 complex, directly demonstrating for the first time that pepducins act through a direct intracellular interaction with their target GPCR. This work established both a mechanistic principle for the pepducin drug class and a general methodology for validating membrane-protein-targeting peptides. 7.4 Epigenetics: Histone Modification Reader and Eraser Identification Post-translational modifications (PTMs) on histones—including methylation, acetylation, phosphorylation, and ubiquitination—regulate chromatin structure and gene expression. The proteins that recognize (“read”) or remove (“erase”) these marks are important targets in cancer epigenetics, but many interactions are weak and transient, evading detection by conventional methods. Yang, Liu, and Li (Chemical Science, 2015) developed diazirine-based peptide probes incorporating photo-leucine to identify histone modification readers and erasers. By placing photo-leucine within histone peptide sequences bearing defined PTMs, the authors created bifunctional probes that bound specifically to the relevant chromatin-associated proteins and crosslinked covalently upon UV irradiation. Coupled with click chemistry enrichment and quantitative LC-MS/MS proteomics, this approach enabled the unbiased identification of reader and eraser proteins from complex nuclear extracts, including multiple previously uncharacterized interactors of specific histone marks. The study established photo-leucine SPPS probes as a powerful platform for epigenomic chemical biology. 7.5 Protein–Polymer Bioconjugation Beyond protein–protein interaction research, photo-leucine chemistry has been exploited in bioconjugation applications for materials science and protein engineering. Lin, Boehnke, and Maynard (Bioconjugate Chemistry, 2014) demonstrated that glutathione S-transferase (GST) metabolically labeled with photo-leucine could be covalently conjugated to glutathione-bearing polymers (polyethylene glycol and polyacrylate derivatives) through UV-mediated crosslinking at the protein–polymer interface. Conjugation occurred specifically at the active site cleft of GST (where glutathione binds), because GST was first pre-bound to its glutathione ligand on the polymer prior to UV irradiation. This affinity-guided photo-crosslinking approach produced protein–polymer conjugates with defined orientation and site-specificity—properties critical for next-generation protein therapeutics and diagnostic biosensors. 7.6 Structural Proteomics and Cross-Linking Mass Spectrometry (XL-MS) Cross-linking mass spectrometry (XL-MS) is an emerging structural biology approach in which chemical crosslinks are used as inter-residue distance restraints to inform or validate structural models of protein complexes. Photo-leucine provides zero-length crosslinks (reporting direct van der Waals contacts within ~3–4 Å), which are complementary to NHS-ester-based crosslinkers that span ~11–12 Å. Iacobucci et al. demonstrated that photo-leucine-labeled trigger factor, a ribosome-associated chaperone, gave 12 UV-crosslinks fully consistent with the published crystal structure, validating the approach for structural proteomics of defined protein systems. The short crosslink length of photo-leucine is particularly valuable for distinguishing interacting surfaces in large protein complexes, where longer crosslinkers can generate ambiguous restraints spanning multiple domains. Integration of photo-leucine crosslink data with cryo-EM density maps provides tight orthogonal restraints for model validation. 7.7 Amyloid and Neurodegeneration Research Photo-leucine has been applied to study the interaction of amyloid β (Aβ) peptide oligomers with neuronal receptors in the context of Alzheimer’s disease. Photo-leucine and photo-methionine were incorporated into model systems to confirm the preferential binding of soluble oligomeric assemblies of Aβ—so-called ADDLs (Aβ-derived diffusible ligands)—with AMPA receptor subunits in neurons. Covalent capture of these transient neurotoxic complexes facilitated their biochemical characterization and provided mechanistic insight into the pathophysiology of amyloid-induced synaptic dysfunction. 7.8 Proteome-Wide Target Profiling in Chemical Proteomics Photo-leucine-containing multifunctional probes—bearing both the photoreactive diazirine and a bioorthogonal enrichment handle (alkyne, azide, or biotin)—enable proteome-wide target identification and profiling in chemical proteomics workflows. In a typical experiment, a diazirine probe is incubated with live cells or cell lysates, UV-crosslinked, and the crosslinked proteins are enriched by streptavidin pulldown (after biotin-azide click chemistry) and identified by quantitative LC-MS/MS. Competitive experiments using the parent compound as competitor confirm on-target enrichment. Cell-based proteome profiling studies using this strategy have identified binding proteins of kinase inhibitors (e.g., dasatinib, staurosporine) and natural product-derived probes in complex cellular proteomes. 8. Comparative Analysis with Alternative Photocrosslinking Agents 8.1 Overview The field of photoaffinity labeling employs three principal classes of photoreactive functional groups: aryl azides, benzophenones, and diazirines (both aliphatic and aryl). A meaningful comparison of these classes across key performance attributes is essential for experimental design. Photo-Leucine (alkyl diazirine) Photophore size: Minimal (3-membered ring) Activation λ (nm): 345–365 Reactive intermediate: Singlet carbene Metabolic incorporation: Yes (mammalian cells) SPPS compatibility: Yes (Fmoc/Boc) UV damage to cells: Low (UV-A) Background labeling: Low–moderate Benzophenone (pBpa) Photophore size: Large (ketone + aryl) Activation λ (nm): 350–365 Reactive intermediate: Triplet diradical Metabolic incorporation: Via amber suppression SPPS compatibility: Yes UV damage to cells: Low (UV-A) Background labeling: Low–moderate Aryl azide Photophore size: Moderate (azide) Activation λ (nm): 250–320 Reactive intermediate: Nitrene / ring-expanded Metabolic incorporation: Via amber suppression SPPS compatibility: Yes UV damage to cells: High (UV-C/B) Background labeling: High Trifluoromethyl diazirine Photophore size: Moderate (CF3 + aryl) Activation λ (nm): 350–360 Reactive intermediate: Carbene Metabolic incorporation: Via amber suppression SPPS compatibility: Yes UV damage to cells: Low (UV-A) Background labeling: Low 8.2 Aryl Azides Aryl azides were among the first photocrosslinking groups explored and offer the advantage of photoactivation at shorter UV wavelengths (250–310 nm). However, this requires UV-B or UV-C irradiation, which is significantly more damaging to cells and biomolecules than UV-A. Upon photolysis, aryl azides generate highly reactive nitrenes and ring-expanded dehydroazepines, which react less selectively and can give higher background labeling. The larger aryl substituent also perturbs structure more than the small aliphatic diazirine. For these reasons, aryl azides have largely been supplanted by diazirines in modern chemical proteomics applications. 8.3 Benzophenone (p-Benzoyl-L-Phenylalanine, pBpa) Benzophenone-containing amino acids such as p-benzoyl-L-phenylalanine (pBpa) react via a triplet diradical mechanism upon irradiation at 350–365 nm, abstracting hydrogen atoms from adjacent C–H bonds. The benzophenone group is larger than the aliphatic diazirine, imposing greater structural perturbation on the peptide or protein in which it is incorporated. Benzophenones cannot be metabolically incorporated by native mammalian aminoacyl-tRNA synthetases and must instead be genetically incorporated via amber suppression technology, requiring cell engineering. They offer the benefit of reversible photochemistry (the triplet diradical can relax back to ground state benzophenone in the absence of an abstractable hydrogen), enabling longer irradiation times and higher crosslinking yields in some applications. However, benzophenones are known to sensitize DNA photodamage, which is a significant concern in cell-based applications. 8.4 Trifluoromethyl Aryl Diazirines 3-Trifluoromethyl-3-phenyldiazirine (the “TPD” photophore) is the most widely used aryl diazirine, offering an improved reactive intermediate profile compared to aryl azides while providing a longer carbene lifetime than aliphatic diazirines. The CF₃ group stabilizes the aryl diazirine thermally while the aryl ring delocalizes the carbene electron density, giving a carbene that is more electrophilic and thus somewhat more selective in its reactivity. The Fmoc-protected aryl diazirine amino acid analogues (e.g., Fmoc-L-Photo-Phe with the TPD photophore) are available commercially. However, the large aryl diazirine substituent introduces greater structural perturbation than the aliphatic methyl diazirine of photo-leucine, and cannot be metabolically incorporated by native translation machinery. Aliphatic diazirines (as in photo-leucine) have been shown to preferentially label acidic surfaces through their diazo intermediate, while aryl diazirines show complementary reactivity, suggesting that combining both probe types may provide more comprehensive interaction mapping. 8.5 Summary of the Advantages of Aliphatic Diazirine (Photo-Leucine) The aliphatic diazirine photophore of photo-leucine offers the optimal combination of: minimal structural perturbation (smallest photophore); metabolic incorporability by native cellular machinery; UV-A activation (least damaging to cells); broad carbene reactivity (captures hydrophobic and other non-nucleophilic interfaces); chemical stability during SPPS; and commercial availability as a protected Fmoc building block. These properties make it the photocrosslinking tool of choice for applications requiring structural mimicry of leucine or incorporation into transmembrane and hydrophobic sequences. 9. Advantages and Limitations 9.1 Key Advantages • Minimal structural perturbation: The 3-methyl-3H-diazirine is the smallest photoreactive group available. Its close mimicry of the leucine isobutyl side chain minimizes disruption to peptide conformation, protein folding, and binding interactions, maximizing the probability that the photo-leucine probe engages its target in the same manner as the parent leucine-containing molecule. • Site-specific incorporation by SPPS: Fmoc-L-Photo-Leucine enables placement of the photophore at any desired position in a synthetic peptide, providing directional, sequence-defined crosslinking with minimal background. • Metabolic incorporation by native machinery: The unprotected form is taken up and incorporated by the mammalian translational apparatus without requiring genetic code expansion or cell engineering, enabling global labeling in living cells. • Broad carbene reactivity: Non-selective insertion into C–H, N–H, and O–H bonds captures interaction interfaces regardless of amino acid composition, including hydrophobic and transmembrane surfaces inaccessible to amine-reactive crosslinkers. • Zero-length crosslinks: Nanosecond carbene lifetime ensures that only molecules in direct van der Waals contact at the moment of photolysis are crosslinked, providing angstrom-resolution spatial information about molecular contacts. • UV-A activation: Irradiation at ~345–365 nm minimally damages DNA, RNA, and proteins, allowing photo-crosslinking in living cells without significant phototoxicity over short irradiation times (1–10 minutes). • Chemical stability: Stable to all Fmoc-SPPS conditions and standard HPLC purification, eliminating the need for special synthetic protocols. • Complementarity with bioorthogonal chemistry: Compatible with probe designs incorporating alkyne or azide handles for click chemistry-based reporter attachment and enrichment. • Broad commercial availability: High-purity material is available from multiple global suppliers, supporting widespread adoption. 9.2 Limitations and Challenges • Moderate crosslinking yields: Productive crosslinking yields are typically 10–40%, limited by aqueous quenching of the carbene, intersystem crossing, and intramolecular rearrangements. This can be a limitation when target proteins are low-abundance or when interaction affinity is very low. • pH-dependent reactivity bias: Recent data demonstrate that alkyl diazirine probes preferentially label acidic amino acid residues (Asp, Glu) and acidic protein surfaces through their diazo intermediate at physiological pH. This bias can lead to overrepresentation of acidic protein patches in crosslinking data and should be considered in interpretation. • Global metabolic labeling lacks sequence precision: When photo-leucine is metabolically incorporated across the entire proteome, crosslinks arise from all leucine positions in all proteins, generating highly complex data that requires advanced MS acquisition strategies and bioinformatics analysis. • No aqueous solubility in Fmoc form: As with all Fmoc-amino acids, organic solvents (DMF, DMSO) are required for dissolution and SPPS, which is standard but adds operational complexity. • UV equipment requirements: Effective photocrosslinking requires a UV-A lamp or LED source with sufficient irradiance at 345–365 nm. Standard laboratory UV transilluminators and gel imagers may not provide optimal wavelength or intensity profiles. • Strict light exclusion required: Ambient light (including fluorescent office lighting, which contains UV-A components) can cause slow photolysis during handling. All work with photo-leucine-containing compounds should be performed under amber-filtered or subdued lighting conditions. • MS complexity of crosslinked peptides: Identification of crosslinked peptide pairs from photocrosslinking experiments by mass spectrometry requires specialized database search algorithms (e.g., pLink, StavroX, MeroX) and complex data analysis workflows not universally available in standard proteomics settings. • No site-specificity in metabolic labeling: Global metabolic incorporation provides no spatial control over where within a protein the photo-leucine is placed, and all leucine positions are labeled stochastically based on local aminoacyl-tRNA synthetase substrate discrimination. 10. Storage and Handling 10.1 Storage Conditions Fmoc-L-Photo-Leucine should be stored at −20°C in a sealed, dry container protected from light. Amber-sealed glass vials or opaque, foil-wrapped containers are recommended. Storage in a desiccator or under inert gas (argon or nitrogen) prevents moisture uptake. Under recommended conditions, commercial lots are stable for at least two years from manufacture. Opened containers should be resealed and returned to cold, dark storage promptly after each use. 10.2 Solution Preparation For SPPS, stock solutions are typically prepared in anhydrous DMF at 0.2–0.5 M concentration immediately before use. For biochemical assays, DMSO solutions (10–100 mM) may be prepared and diluted into aqueous buffer; the maximum DMSO concentration in the final assay mixture should be kept below 1% v/v to avoid protein denaturation. All solutions should be prepared and stored in amber vials on ice, with minimal exposure to ambient light. 10.3 Light Management All handling of Fmoc-L-Photo-Leucine and downstream products (peptides, probes, labeled proteins) should be performed in amber-filtered or low-UV laboratory lighting. Standard yellow “safe lights” used in photographic darkrooms or commercial amber LED lighting are suitable alternatives. HPLC purification should use UV detection wavelengths below 280 nm, avoiding the 345 nm diazirine absorption band, and purified fractions should be collected in amber tubes. 11. Representative Experimental Protocols 11.1 Incorporation into Synthetic Peptides by Fmoc-SPPS • Prepare a 0.2 M solution of Fmoc-L-Photo-Leucine in anhydrous DMF immediately before use; protect from light. • Load Fmoc-SPPS resin (e.g., Rink amide MBHA, Wang) and perform amino acid couplings using 4–5 equivalents of each Fmoc-amino acid, activated with HATU (3.8 equiv) and DIPEA (8 equiv) in DMF (3 mL per gram resin), 20–45 minutes per coupling step. • Couple Fmoc-L-Photo-Leucine at the desired position exactly as other Fmoc-amino acids. Double coupling may be used for difficult positions. • Deprotect Fmoc with 20% piperidine in DMF (1 × 5 min, 1 × 10 min); wash thoroughly with DMF. • Continue chain assembly; all subsequent steps are performed under amber lighting. • Cleave from resin with TFA/TIS/H₂O (95:2.5:2.5, v/v/v) for 2–4 hours at RT in amber vials. Precipitate crude peptide with ice-cold diethyl ether (×3). • Purify by reverse-phase HPLC (C18 column; 5–65% acetonitrile/0.1% TFA gradient; UV detection at 214 nm) under amber lighting. Collect fractions in amber tubes; verify by MALDI-TOF or ESI-MS. • Confirm diazirine integrity in purified peptide by UV absorbance at 345 nm. Lyophilize and store at −20°C protected from light. 11.2 Metabolic Labeling and UV Photocrosslinking in Mammalian Cells • Grow cells (e.g., HeLa, HEK293, COS7) to 60–70% confluency in standard culture medium. • Wash cells ×2 with PBS; replace with Leu/Ile/Val-deficient and Met-deficient DMEM supplemented with 10% dialyzed FBS. Incubate 45–60 min to deplete intracellular amino acid pools. • Add photo-leucine (4 mM) and photo-methionine (1.7 mM) to the deficient medium. Incubate 22 hours at 37°C, 5% CO₂. • Remove labeling medium; wash cells ×2 with ice-cold PBS. Add 1–2 mL PBS and transfer to ice (keep plate horizontal). • Irradiate cells with UV-A lamp (320–365 nm; 200 W mercury lamp with 310 nm cutoff filter, or UV-A LED array) from 1–5 cm above the cells for 1–10 min on ice. Include non-irradiated controls. • Lyse cells in RIPA buffer or appropriate lysis buffer. Clarify by centrifugation. Proceed to western blot, immunoprecipitation, or mass spectrometry analysis. 11.3 Photoaffinity Target Identification with a Synthetic Probe • Synthesize a bifunctional photoaffinity probe: incorporate Fmoc-L-Photo-Leucine at the desired position and include an alkyne handle (e.g., homopropargylglycine) at a distal, non-functional position. Purify and confirm by LC-MS. • Validate probe activity (e.g., binding assay, cell-based functional assay) to confirm that the photo-leucine substitution does not ablate biological activity. • Incubate probe (1–10 µM) with cell lysate, membrane fraction, or purified protein at 4°C in amber tubes for 30–60 min to allow equilibrium binding. • Transfer to ice; UV-irradiate (345–365 nm, 10 min). Include a non-irradiated control, a dark (no UV) control, and a competition control (excess parent compound + probe + UV). • Perform CuAAC click chemistry: add azide–biotin (or azide–rhodamine) (100 µM), CuSO₄ (1 mM), sodium ascorbate (1 mM), TBTA ligand (0.1 mM) in PBS; react 1 h at RT. • Enrich biotinylated proteins with streptavidin-coated magnetic beads (1 h, 4°C, rotating). Wash stringently (×5 with RIPA buffer). • On-bead trypsin digestion; analyze by LC-MS/MS with label-free or isobaric (TMT/iTRAQ) quantification. Compare probe-treated vs. competition-treated samples to identify specific targets. 12. Recent Advances and Emerging Directions 12.1 Mechanistic Elucidation: The Diazo Intermediate A significant advance in understanding diazirine photochemistry was reported in Nature Communications (2024), revealing that alkyl diazirine photolysis proceeds sequentially through a diazo intermediate before generating the free carbene. This two-step mechanism has important implications for probe design and data interpretation. The diazo intermediate preferentially targets polar, buried residues—especially acidic residues (Asp, Glu) inaccessible to conventional amine-reactive crosslinkers—through a relatively selective electrophilic mechanism, while the subsequent carbene provides the broad, non-selective crosslinking for which diazirines are known. By tuning irradiation wavelength and intensity, researchers can now bias the crosslinking reaction toward diazo-mediated specificity or carbene-mediated non-selectivity, enabling complementary structural information from a single probe. 12.2 Development of Next-Generation Diazirine Tags Several groups have reported engineered diazirine photophores with improved properties for specific applications. West and Woo (JACS, 2022) developed PALBOX, a cyclobutane diazirine-based photoaffinity tag with reduced pH-dependent reactivity compared to standard alkyl diazirines. PALBOX probes show differential reactivity profiles and reduced labeling of off-target acidic proteins, potentially improving selectivity in chemoproteomic applications. These advances in diazirine engineering are expected to inform the design of next-generation photo-amino acid building blocks with improved target selectivity. 12.3 Integration with Quantitative Chemical Proteomics The combination of photo-leucine photocrosslinking with isobaric labeling quantitative proteomics (TMT, iTRAQ, or isotope-labeled analogue competitive probes in the ABPP-SILAC framework) is enabling proteome-wide, quantitative mapping of drug and probe interactions across entire cellular proteomes. These workflows identify both primary targets and off-targets of bioactive molecules with statistical rigor, supporting both early drug discovery and clinical pharmacology. 12.4 Spatial Proteomics and Compartment-Specific Crosslinking Photo-leucine probes are being integrated with spatial proteomics strategies that use organelle-targeted peroxidases (APEX2) or BioID proximity labeling enzymes to first define the local protein neighborhood, followed by photo-leucine crosslinking to covalently capture direct interactions within that neighborhood. This layered approach combines the sensitivity of proximity labeling (for detecting low-abundance neighbors) with the specificity of photo-crosslinking (for confirming direct contact). 12.5 Applications in Structural Glycobiology Photo-leucine and related diazirine amino acids are increasingly being applied in combination with glycopeptide probes to study glycoprotein–protein interactions. Tanaka and Kohler demonstrated the metabolic incorporation of diazirine-modified sugars to study glycan-protein contacts; analogous strategies are being developed combining photo-leucine incorporation in protein backbones with glycoengineering to map the proteome of specific glycan-decorated cell surface microenvironments. 12.6 Therapeutic Probe Design: Covalent Photo-Drugs The concept of photoactivatable covalent drugs—molecules that specifically crosslink to their target only upon light activation—is an emerging frontier in targeted therapy. Photo-leucine-containing peptidomimetics and stapled peptides are being explored as candidates for photodynamic cancer therapy, where the UV or visible light activates covalent target engagement only in the irradiated tumor volume, reducing systemic off-target toxicity. This application remains early-stage but represents a compelling future direction for the chemistry pioneered by photo-leucine. 13. Conclusions and Outlook Fmoc-L-Photo-Leucine stands as one of the most versatile and impactful tools in the modern chemical biologist’s toolkit. Its unique combination of structural minimality—achieved through the smallest available photophore—with the power of covalent photocrosslinking chemistry has enabled discoveries ranging from novel protein–protein interactions and drug target identifications to GPCR mechanism elucidation and site-specific protein–polymer bioconjugation. The compound’s value is amplified by the dual modalities it enables: metabolic global labeling in living cells (using the unprotected form) and rational, site-specific probe design through Fmoc-SPPS (using the Fmoc-protected form). Together, these two strategies cover the full spectrum from unbiased, hypothesis-free interactome mapping to targeted, hypothesis-driven structural and mechanistic studies. No other photoreactive amino acid currently combines both capabilities as effectively. Looking forward, several developments are poised to further expand the impact of photo-leucine chemistry. The mechanistic clarification of the diazo intermediate pathway opens new avenues for tuneable, selectivity-controlled crosslinking. The development of next-generation engineered diazirine photophores with improved selectivity (e.g., PALBOX) will be readily translatable to photo-amino acid scaffolds. Integration with advanced quantitative proteomics, spatial biology, and structural cryo-EM workflows will continue to elevate the scientific value of photo-crosslinking data. And emerging applications in therapeutic photo-covalent probe design represent an exciting frontier where the chemistry of photo-leucine may ultimately contribute to the development of new medicines. Fmoc-L-Photo-Leucine thus remains not merely a reagent of historical interest but an actively developing and increasingly central tool for interrogating the molecular basis of life in health and disease—a compound whose full potential has yet to be realized. References 1. Suchanek M, Radzikowska A, Thiele C. Photo-leucine and photo-methionine allow identification of protein–protein interactions in living cells. Nat Methods. 2005;2(4):261–268. DOI: 10.1038/nmeth752. [Original description of photo-leucine technology] 2. MacKinnon AL, Garrison JL, Hegde RS, Taunton J. Photo-leucine incorporation reveals the target of a cyclodepsipeptide inhibitor of cotranslational translocation. J Am Chem Soc. 2007;129(47):14560–14561. DOI: 10.1021/ja076250y. [Improved synthesis and drug target ID] 3. Vila-Perelló M, Pratt MR, Tulin F, Muir TW. Covalent capture of phospho-dependent protein oligomerization by site-specific incorporation of a diazirine photo-cross-linker. J Am Chem Soc. 2007;129(27):8068–8069. DOI: 10.1021/ja072013j. [Semi-synthetic protein with photo-Met; EPL approach] 4. Janz JM, Ren Y, Looby R, et al. Direct Interaction between an Allosteric Agonist Pepducin and the Chemokine Receptor CXCR4. J Am Chem Soc. 2011;133(39):15878–15881. DOI: 10.1021/ja206661w. [GPCR-pepducin interaction; SPPS photo-Leu] 5. Lin EW, Boehnke N, Maynard HD. Protein-Polymer Conjugation via Ligand Affinity and Photoactivation of Glutathione S-Transferase. Bioconjug Chem. 2014;25(10):1902–1909. DOI: 10.1021/bc500380r. [Protein-polymer bioconjugation] 6. Yang T, Liu Z, Li XD. Developing diazirine-based chemical probes to identify histone modification ‘readers’ and ‘erasers’. Chem Sci. 2015;6(2):1011–1017. DOI: 10.1039/C4SC02328E. [Epigenetics application] 7. van der Meijden B, Robinson JA. Synthesis of a polymyxin derivative for photolabeling studies in the gram-negative bacterium Escherichia coli. J Pept Sci. 2015;21(3):231–235. DOI: 10.1002/psc.2736. [Antibiotic target ID] 8. Iacobucci C, et al. Protocol for High-Yield Production of Photo-Leucine-Labeled Proteins in Escherichia coli. J Proteome Res. 2020;19(10):4071–4081. DOI: 10.1021/acs.jproteome.0c00105. [High-yield E. coli expression] 9. Das J. Aliphatic diazirines as photoaffinity probes for proteins: recent developments. Chem Rev. 2011;111(8):4405–4417. DOI: 10.1021/cr1002722. [Comprehensive diazirine review] 10. Hill JR, Robertson AAB. Fishing for Drug Targets: A Focus on Diazirine Photoaffinity Probe Synthesis. J Med Chem. 2018;61(16):6945–6963. DOI: 10.1021/acs.jmedchem.7b01561. [Synthetic focus review] 11. Homan EA, et al. Photoaffinity labelling with small molecules. Nat Rev Methods Primers. 2024;4:30. DOI: 10.1038/s43586-024-00308-4. [Comprehensive PAL primer] 12. Zhang Z, et al. Dissecting diazirine photo-reaction mechanism for protein residue-specific cross-linking and distance mapping. Nat Commun. 2024;15:6083. DOI: 10.1038/s41467-024-50315-y. [Mechanistic study; diazo intermediate] 13. West AV, Woo CM. Design and Evaluation of a Cyclobutane Diazirine Alkyne Tag for Photoaffinity Labeling in Cells. J Am Chem Soc. 2022;144(46):21174–21183. DOI: 10.1021/jacs.2c08257. [Next-gen diazirine PALBOX] 14. Halloran MW, Lumb JP. Recent applications of diazirines in chemical proteomics. Chem Eur J. 2019;25(20):4885–4898. DOI: 10.1002/chem.201806072. [Chemical proteomics review] |
|
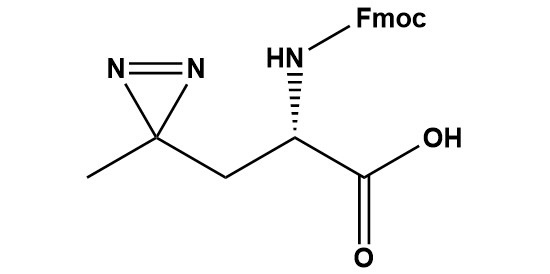
Fmoc-Photo-Leu-OH
For Research & Development use only. Not for testing and/or use on humans.
