
Contents
1. Resins for Boc SPPS
1.1. Merrifield Resin (Chloromethylpolystyrene)
1.2. PAM Resin
1.3. BHA Resin
1.4. MBHA Resin
2. The Boc Group
2.1. General Remarks
2.2. Deprotection
2.3. Neutralization
3. Cleavage from the Resin
3.1. HF
3.2. TFMSA
3.3. TMSOTf
3.4. HBr/TFA
4. Side Reactions
4.1. Diketopiperazine Formation
4.2. Aspartimide Formation
4.3. Homoserine Lactone Formation
4.4. N-O Shift
4.5. Side Reaction Involving Glu
4.6. Asp-Pro Cleavage
5. References
6. Related Products
7. Useful Links

1.1 Merrifield Resin (Chloromethylpolystyrene)
It is the most classical resin used in Boc based SPPS. The first amino acid is introduced as a salt which insures a racemization free esterification. In the original procedure [1] the triethylammonium salt of the Boc-amino acid was used but the formation of quaternary triethylammonium salt between the chioromethylated resin and the triethylamine limits the usefulness of the method. Presently the Boc-amino acids are esterified via their cesium salt, following the method of Gisin [2].
The deprotection (cleavage of the Boc group) is realized using a 50% solution of TFA in DCM.
The benzyl ester linkage between the C-terminal amino acid and the polystyrene can be partially cleaved in the deprotection step and important losses can occur during the construction of a long peptide chain [3].
1.2. PAM Resin
To circumvent these losses a phenylacetamido linker has been inserted between the support and the C-terminal amino acid. The stabilization introduced by this linker reduces the loss of peptide at each deprotection step and the peptide acid is released from the resin by HF [4,5].
For the preparation of the Boc-amino acid-PAM resin, the preformed Boc-amino acid-PAM handle is coupled to an aminomethyl resin. Indeed the esterification of the Boc-amino acid to the hydoxymethyl-phenylacetamidomethylpolystyrene using DCC/DMAP implies a risk of racemization.
In addition this last procedure leaves unreacted hydroxyl groups on the resin and it is advisable to cap them by acetylation [6] or benzoylation [7].
For both types of support synthesis can be successfully performed with resins having a substitution in the range of 0.5 to 1.0 mmol/g. However certain sequences are more conveniently prepared with a resin of lower substitution.
1.3. BHA Resin
The BHA-resin has been introduced by Pietta and Marshall [10] for the preparation of peptide amides.
However it was rapidly recognized that the stability of the amide bond between the C-terminal amino acid and the resin depends on the amino acid. The enhanced stability can prevent an effective cleavage of the peptide from the resin.
Orlowsky [11] has developed the more Iabile 4-methoxybenzhydrylamine resin but in this case undesirable losses of peptide have been encountered during the TFA cleavage of the Boc group.
1.4. MBHA Resin
This resin has been introduced by Stewart and Matsueda [12] and shows an optimal balance between stability towards TFA and lability towards HF. The MBHA resin has become the support of choice for the preparation of peptide amides in the Boc SPPS.
The coupling of the first amino acid is realized using a standard coupling protocol (DCC/HOBt, HBTU, TBTU, …).
A substitution level between 0.5 and 1.0 mmol/g is routinely used as mentioned above but the optimal load for a specific sequence can only be determined after comparison of test syntheses.
2. The Boc Group
2.1. General Remarks
The Boc group has been described by Carpino in 1957 [13] and has directly been applied in peptide synthesis.
Merrifield has rapidly realized that the relative labilities towards acid of the Boc and benzyl groups would be compatible with an effective temporary vs. permanent protecting scheme and very soon published the solid phase synthesis of Bradykinin using Boc protected amino acids [14].
2.2. Deprotection
In most cases the cleavage of the Boc group is performed with a 50% TFA/DCM solution. A short (5 minutes) prewash is made followed by the cleavage reaction (15 to 25 minutes). The use of a scavenger is recommended when Cys, Met or Trp are present in the sequence. In that case 0.5% DTE are added to the TFA/DCM solution.
Neat TFA has also been used for the cleavage of the Boc group [15] but this protocol is rarely used.
After the cleavage, the peptide-resin is washed with DCM followed by shrinking with an alcohol to “extract” most of the remaining TFA.
IPA is preferred to MeOH because its use reduces the risk of transesterification with the Asp/Glu side chain protecting groups, and also because of a lower toxicity.
RECOMMENDED STANDARD PROCEDURE
Boc Cleavage
The peptide-resin is swollen with DCM. The peptide-resin is stirred in 50% TFA/DCM for 5 minutes (10 mL/g peptide-resin). After filtration, a fresh solution of 50%TFA/DCM is introduced and the peptide resin is stirred for an additional 20 minutes. When Cys, Met and Trp are present, 0.5% DTE is added to the TPA solution. The peptide-resin is washed with DCM (2x) and IPA (2x) before neutralization.
|
2.3. Neutralization
The deprotection reaction leaves the amino group as a trifluoroacetate. It is necessary to neutralize that salt before the coupling,
This is realized in two steps in which the peptide-resin is suspended and stirred in a 10% TEA/DCM solution. The resin is then carefully washed before proceeding to the coupling step.
RECOMMENDED STANDARD PROCEDURE
Neutralization
The peptide-resin is suspended and stirred in 10% TEA/DCM (10 mL/g peptide-resin) for 2×10 minutes. After filtration it is washed alternately with DCM and IPA (3xeach) before being washed with the solvent used for the coupling.
|
3. Cleavage from the Resin
In this last step of the synthesis the peptide is cleaved from the resin together with the side chain protecting groups at the exception of the ones which are orthogonal to this acidolytic reaction.
Among these, two must be cleaved from the amino acid before the final step: Dnp from His and For from Trp.
RECOMMENDED STANDARD PROCEDURE
Dnp Cleavage
The peptide-resin is swollen in DMF and a 20 fold excess of thiophenol in DMF is added.
|
RECOMMENDED STANDARD PROCEDURE
For Cleavage
A solution of 10% piperidine in DMF is cooled in an ice bath.
|
3.1. HF
Liquid hydrogen fluoride is without any doubt the most commonly used reagent for the cleavage of the peptide from the resin, with concomitant cleavage of the side-chain protecting groups [16].
The main drawback of the HF treatment resides in its handling as it is indeed extremely toxic, corrosive and volatile.
A special all-fluorocarbon apparatus is required for reactions involving HF since it reacts with glass.
Such equipment is commercially available and it is recommended to follow precisely the manufacturer’s instructions in view of the risk represented by the use of HF.
The limited availability of this special equipment in large volume units prevents the scale-up of the procedure.
The cleavage reaction is usually performed for 45 to 60 minutes at a temperature close to 0°C but can be prolonged if several Arg(Tos) residues are present in the sequence.
Due to the high concentration of alkylating species in the reaction medium, it is indispensable to include scavengers in the cleavage mixture. Anisole has been widely used in a ratio HF/anisole 9:1, but sulfur containing scavengers such as thiocresol and dimethyl sulfide (DMS) are also recommended.
At the end of the reaction the HF is evaporated under vacuum and the peptide extracted from the resin and isolated.
RECOMMENDED STANDARD PROCEDURE
HF Cleavage
The peptide-resin is placed in the reactor equipped with a teflon coated stirring bar.
|
Besides the standard HF procedure, the low-high HF cleavage protocol of Tam and Merrifield [17] consists of a two-steps procedure.
In the first step, a low concentration of HF in DMS (1:3) cleaves most of the protecting groups following an SN2 mechanism (vs. SN1 under the standard conditions). The formation of carbocations and most of the side reactions are prevented.
After evaporation of HF and DMS, a high HF cleavage is performed to cleave the functionalities resistant to the low HF conditions such as Arg(Tos).
The large volumes of DMS and the extra time needed to evaporate the first DMS/HF mixture represent the main drawbacks of the method.
RECOMMENDED STANDARD PROCEDURE
Low-High HF Cleavage
The peptide-resin is placed in the reactor together with DMS/p-cresol
|
3.2. TFMSA
TFMSA can be used as an alternative for HF [18]. The main advantage of TFMSA is that glass is inert towards this acid and normal laboratory glassware can be used for the cleavage.
On the other hand, TFMSA will not cleave Arg (Tos) or Arg (NO2). Also, Asp(OcHex) and Cys(MBz1) are not cleaved by TFMSA.
In addition TFMSA will not cleave the pep- tide from BHA-resin and requires prolonged reaction time for the cleavage from MBHA-resin.
In view of those potential problems it is recommended to optimize the reaction conditions performing small scale orientation cleavages.
RECOMMENDED STANDARD PROCEDURE
TFMSA Cleavage
The peptide-resin is placed in a round-bottom flask with a stirring bar.
|
3.3. TMSOTf
TMSOTf represents an alternative to HF [19]. The cleavage of Arg(Tos) requires a longer reaction time.
Cys(Bzl), Cys(Acm) and Arg(NO2) are stable to TMSOTf treatment.
A two step cleavage procedure has also been developed using TMSBr/thioanisole/TFA in the first step (cleavage of the protecting groups) and TMSOTf/thioanisole/TFA in the second step (cleavage from the resin) [20].
3.4. HBr/TFA
HBr/TFA has been used for the cleavage of the peptide resin linkage from the first days of the solid phase technique [21]; however the reagent has shown severe limitations and has been left aside since the introduction of HF.
A special mixture (HBr/TFA/pentamethylbenzene/thioanisole) has been developed which allows the cleavage of a peptide amide from MBHA resin [22] but the general benefits of this method have not been demonstrated.
4. Side Reactions
The repetitive TFA treatments together with the final HF cleavage represent sources of side-reactions for the most sensitive amino acids such as Cys, Met and Trp.
Alkylation by carbocations generated in the acidolytic cleavage is the most prominent side- reaction but the judicious choice of scavengers will help to minimize unwanted modifications.
4.1. Diketopiperazine Formation
The extent of this side-reaction can be reduced compared to the situarion in Fmoc based SPPS Indeed, after the deprotection reaction in Boc SPPS the n-amino function is protonated and less prone to attack the benzyl ester bond.
Protocols have been developed in which the neutralization step is realized concomitantly with the coupling [23,24] to suppress this side- reaction.
4.2. Aspartimide Formation
Aspartimide formation can be a source of problems, particularlyin peptides containing the Asp-Gly, Asp-Ala or Asp-Ser sequences.
4.3. Homoserine Lactone Formation
C-terminal Met can cyclize to homoserine lactone [25] during the HF cleavage if t-butyl type protecting groups have not been cleaved before the HF treatment.

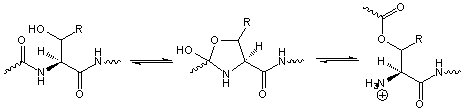
4.4. N-O Shift
N-O shift can occur during the treatment with strong acid of peptides containing Ser or Thr [26] but the reaction can be reversed by base treatment.
4.5. Side Reaction Involving Glu
The γ carboxyl function of Glu, protonated in strong acid, can lose water and form an acylium ion which will cyclize to a pyrrolidone or be trapped by a scavenger such as anisole to yield a ketone [27].

4.6. Asp-Pro Cleavage
The cleavage of the Asp-Pro bond has been observed during HF treatment [28].
5. References
[1] R.B. Merrifield, Biochemistry 3 (1964) 1385.
[2] BR Gisin, Helv. Chim. Acta 56 (1973) 1476.
[3] A.R. Mitchell, B.W. Erickson, M.N. Ryabtsev, RS. Hodges, R.B. Merrifield,J. Am. Chem. Soc. 98 (1976) 7357.
[4] AR. Mitchell, S.B.H. Kent, M. Engeihard, R.B. Merrifield,J. Org. Chem. 43 (1978) 2845.
[5] J.P. Tam, S.B.H. Kent, T.W. Wong, R.B. Merrifield, Synthesis (1979) 955.
[6] R.B. Merrifield in The Chemistry ofPolypeptides. P.G. Katsoyannis Ed. Plenum, New York 1973,336.
[7] S.S. Wang,J. Org. Chem. 40 (1975) 1235.
[8] M. Manning,J. Am. Chem. Soc. 90 (1968)1348.
[9] J.M. Stewart,J.D. Young, Solid Phase Peptide Synthesis, 2nd Edition, Pierce Chemical Company, Rockford 111984, 44.
[10] P.G. Pietta, G.R. Marshall,J. Chem. Soc., Chem. Commun. (1970) 650.
[11] R.C. Orlowski, R. Walter, D. Winkler,J. Org. Chem. 41 (1976) 3701.
[12] G.R. Matsueda,J.M. Stewart, Peptides 2(1981) 45.
[13] L.A. Carpino,J. Am. Chem. Soc. 79(1957) 4427.
[14] RB. Merrifield, Biochemistry 3 (1964) 1385.
[15] S.B.H. Kent, D. Alewood, P. Alewood, M. Baca, A. Jones, M. Schnolzer in 2nd International
Symposium on Innovation and Perspectives in SPPS Canterbury 1991, R. Epton Ed., Intercept Ltd, Andover 1992, 1.
[16] S. Sakakibara, Y. Shimonishi, Y. Kishida, M. Okada, H. Sugihara, Bull. Chem. Soc. Jpn. 40 (1967) 2164
[17] J.P. Tam, W.F. Heath, R.B. Merrifield,J. Am.Chem. Soc. 105 (1983) 6442.
[18] H. Yajima, N. Fujii, H. Ogawa, H. Kawatani,J. Chem. Soc., Chem. Commun. (1974) 107.
[19] N. Fujii, A. Okata, 0. Ikemura, K. Akaji, S. Funakoshi, Y. Hayashi, Y. Kuroda, H. Yajima, J. Chem. Soc., Chem. Commun. (1987) 274.
[20] M. Nomizu, Y. Anagaki, T. Yamashita, A. Ohkubo, A. Otaka, N. Fujii, P.P. Roller, H. Yajima, Int.J. Pept. Protein Res. 37(1991)145.
[21] G.R Marshall, RB. Merrifield, Biochemistry 4 (1965) 2394.
[22] S.S. Wang, B.S.H.Wang,J.L. Hughes, Ej. Leopold, C.R Wu,J.P. Tam, Int.J. Pept. Protein Res. 40 (1992) 344.
[23] D.L. Nguyen, A. Heitz, B. Castro,J. Chem. Soc., Perkin Trans. 1 (1987)1915.
[24] M. Gain, P. Lloyd-Williams, F. Albericio, E. Giralt, Tetrahedron Lett. 31 (1990) 7363.
[25] M. Gairl, P. Lloyd-Williams, F. Albericio, E. Giralt, Tetrahedron Lett. 35 (1994) 175.
[26] S. Sakakibara, K.H. Shin, G.P. Hess,J. Am. Chem. Soc. 84(1962)4921.
[27] R.S. Feinberg, RB. MerrifIeld,J. Am. Chem. Soc. 97(1975) 3485.
[28] C.R Wu,J.D. Wade, G.W. Tregear, Int.J. Pept. Protein Res. 31 (1988) 47.
7. Useful Links
Peptide Synthesis
Introduction to Peptide Synthesis
In Situ Neutralization in Boc-chemistry Solid Phase Peptide Synthesis
Anhydrous Hydrogen Fluoride Cleavage in Boc Solid Phase Peptide Synthesis
Synthesis of peptides using tert-butyloxycarbonyl (Boc) as the α-amino protection group
Methods and protocols of modern solid phase Peptide synthesis
Linkers, resins, and general procedures for solid-phase peptide synthesis
Peptides, solid-phase synthesis and characterization: Tailor-made methodologies
Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences
Fmoc solid phase peptide synthesis