
20 Amino Acids
This entry is from Wikipedia, the leading user-contributed encyclopedia.
|
• Alanine |
• Cysteine |
• Histidine |
• Methionine |
• Threonine |
|
|
• Arginine |
• Glutamic Acid |
• Isoleucine |
• Phenylanine |
• Tryptophan |
|
|
• Asparagine |
• Glutamine |
• Leucine |
• Proline |
• Tyrosine |
|
|
• Aspartic Acid |
• Glycine |
• Lysine |
• Serine |
• Valine |
Alanine
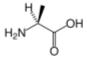
Alanine is an α-amino acid with the chemical formula HO2CCH(NH2)CH3. The L-isomer is one of the 20 proteinogenic amino acids, i.e. the building blocks of proteins. Its three letter code is ala, its one letter code is A, and its codons are GCU, GCC, GCA, and GCG.[1] It is classified as an nonpolar amino acid. L-alanine is second only to leucine, accounting for 7.8% of the primary structure in a sample of 1,150 proteins [1]. D-alanine occurs in bacterial cell walls and in some peptide antibiotics.
1. Structure
The α-carbon atom of alanine is bound with a methyl group (-CH3), making it one of the simplest α-amino acids with respect to molecular structure and also resulting in alanine being classified as an aliphatic amino acid. The methyl group of alanine is non-reactive and is thus almost never directly involved in protein function.
2. Biosynthesis
Alanine is most commonly produced by reductive amination of pyruvate. Because transamination reactions are readily reversible and pyruvate pervasive, alanine can be easily formed and thus has close links to metabolic pathways such as glycolysis, gluconeogenesis, and the citric acid cycle. It also arises together with lactate and generates glucose from protein via the alanine cycle.
3. Sources
Any protein-containing food such as meat, poultry, fish, eggs or dairy products is rich in alanine. Racemic alanine can be prepared via the addition of hydrogen cyanide and ammonia to acetaldehyde by the Strecker reaction.[2]
4. References
1. Doolittle RF (1989). “Redundancies in protein sequences” in Prediction of Protein Structures and the Principles of Protein Conformation. (Fasman GD, ed.), pp 599-623, Plenum Press, New York.
2. IUPAC-IUBMB Joint Commission on Biochemical Nomenclature. Nomenclature and Symbolism for Amino Acids and Peptides. Recommendations on Organic & Biochemical Nomenclature, Symbols & Terminology etc.
3. Kendall, E. C.; McKenzie, B. F. “dl-Alanine” Organic Syntheses, Collected Volume 1, p.21 (1941).
Arginine
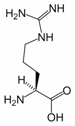
Arginine (symbol Arg or R) is an α-amino acid. The L-form is one of the 20 most common natural amino acids. In mammals, arginine is classified as a semiessential or conditionally essential amino acid, depending on the developmental stage and health status of the individual. Infants are unable to effectively synthesize arginine, making it nutritionally essential for infants. Adults, however, are able to synthesize arginine in the urea cycle.
Arginine was first isolated from a lupin seedling extract in 1886 by the Swiss chemist Ernst Schulze.
Contents
1. Structure
2. Synthesis
3. Functions
3.1. In proteins
3.2. As a precursor
3.3. Implication in herpes simplex viral replication
3.4. Implication in contributing to risk of death from heart disease
4. Food sources
5. References
1. Structure
Arginine can be considered to be a basic amino acid because the part of the side chain nearest to the backbone is long, carbon-containing and hydrophobic, whereas the end of the side chain is a complex guanidinium group. With a pKa of 12.48, the guanidinium group is positively charged in neutral, acidic and even most basic environments. Because of the conjugation between the double bond and the nitrogen lone pairs, the positive charge is delocalized. This group is able to form multiple H-bonds.
2. Synthesis
Arginine is synthesized from citrulline by the sequential action of the cytosolic enzymes argininosuccinate synthetase (ASS) and argininosuccinate lyase (ASL). This is energetically costly, as the synthesis of each molecule of argininosuccinate requires hydrolysis of adenosine triphosphate (ATP) to adenosine monophosphate (AMP); i.e., two ATP equivalents.
Citrulline can be derived from multiple sources:
• from arginine via nitric oxide synthase (NOS);
• from ornithine via catabolism of proline or glutamine/glutamate;
• from asymmetric dimethylarginine (ADMA) via DDAH.
The pathways linking arginine, glutamine, and proline are bidirectional. Thus, the net utilization or production of these amino acids is highly dependent on cell type and developmental stage.
On a whole-body basis, synthesis of arginine occurs principally via the intestinal–renal axis, wherein epithelial cells of the small intestine, which produce citrulline primarily from glutamine and glutamate, collaborate with the proximal tubule cells of the kidney, which extract citrulline from the circulation and convert it to arginine, which is returned to the circulation. Consequently, impairment of small bowel or renal function can reduce endogenous arginine synthesis, thereby increasing the dietary requirement.
Synthesis of arginine from citrulline also occurs at a low level in many other cells, and cellular capacity for arginine synthesis can be markedly increased under circumstances that also induce iNOS. Thus, citrulline, a coproduct of the NOS-catalyzed reaction, can be recycled to arginine in a pathway known as the citrulline-NO or arginine-citrulline pathway. This is demonstrated by the fact that in many cell types, citrulline can substitute for arginine to some degree in supporting NO synthesis. However, recycling is not quantitative because citrulline accumulates along with nitrate and nitrite, the stable end-products of NO, in NO-producing cells.[1]
3. Functions
Arginine plays an important role in cell division, the healing of wounds, removing ammonia from the body and the immune function. Arginine, taken in combination with pycnogenol[2] or yohimbine[3], has also been used as a treatment for erectile dysfunction.
3.1. In proteins
The geometry, charge distribution and ability to form multiple H-bonds make arginine ideal for binding negatively charged groups. For this reason arginine prefers to be on the outside of the proteins where it can interact with the polar environment. Incorporated in proteins, arginine can also be converted to citrulline by PAD enzymes. In addition, arginine can be methylated by protein methyltransferases.
3.2. As a precursor
Arginine is the immediate precursor of NO, urea, ornithine and agmatine; is necessary for the synthesis of creatine; and can not be used for the synthesis of polyamines (mainly through ornithine and to a lesser degree through agmatine), citrulline, and glutamate. For being a precursor of NO, (relaxes blood vessels), arginine is used in many conditions where vasodilation is required. The presence of asymmetric dimethylarginine (ADMA), a close relative, inhibits the nitric oxide reaction; therefore, ADMA is considered a marker for vascular disease, just as L-arginine is considered a sign of a healthy endothelium.
3.3. Implication in herpes simplex viral replication
Tissues culture studies have shown the suppression of viral replication when the lysine to arginine ratio in vitro favors lysine. The therapeutic consequence of this finding is unclear, but dietary arginine may affect the effectiveness of lysine supplementation.[4]
3.4. Implication in contributing to risk of death from heart disease
A recent Johns Hopkins study testing the addition of L-arginine to standard postinfarction treatment has implicated L-arginine supplementation with an increased risk of death in patients recovering from heart attack.[5] This study has been discussed in some detail in : “Reverse Heart Disease Now” by Stephen T Sinatra MD and James C Roberts MD, publ. Wiley 2006 ISBN 0-471-74704-1 at pp 111 -113.
4. Food sources
Arginine is found in chocolate, wheat germ and flour, buckwheat, granola, oatmeal, dairy products (cottage cheese, ricotta, nonfat dry milk, skim yogurt), beef (roasts, steaks), pork (Canadian bacon, ham), nuts (coconut, pecans, cashews, walnuts, almonds, Brazil nuts, hazel nuts, peanuts), seeds (pumpkin, sesame, sunflower), poultry (chicken and turkey light meat), wild game (pheasant, quail), seafood (halibut, lobster, salmon, shrimp, snails, tuna in water), chick peas, cooked soybeans, and some energy drinks.
5. References
1. Enzymes of arginine metabolism J Nutr. 2004 Oct; 134(10 Suppl): 2743S-2747S.
2. Stanislavov, R. and Nikolova. 2003. Treatment of Erectile Dysfunction with Pycnogenol and L-arginine. Journal of Sex and Marital Therapy, 29(3): 207-213.
3. Lebret, T., Hervéa, J. M., Gornyb, P., Worcelc, M. and Botto, H. 2002. Efficacy and Safety of a Novel Combination of L-Arginine Glutamate and Yohimbine Hydrochloride: A New Oral Therapy for Erectile Dysfunction. European Urology 41(6): 608-613.
4. Griffith RS, Norins AL, Kagan C. (1978). “A multicentered study of lysine therapy in Herpes simplex infection”. Dermatologica. 156 (5): 257-267.
5. Arginine Therapy in Acute Myocardial Infarction JAMA. 2006 Jan; Vol.295 #1: 58-64.
Asparagine
Asparagine is one of the 20 most common natural amino acids on Earth. It has carboxamide as the side chain’s functional group. It is considered a non-essential amino acid.
Its three-letter abbreviation is Asn, and its one-letter abbreviation is N. A three-letter designation for either asparagine or aspartic acid is Asx (one-letter abbreviation: B).
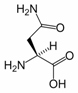
A reaction between asparagine and reducing sugars or reactive carbonyls produces acrylamide (acrylic amide) in food when heated to sufficient temperature, i.e. baking. These occur primarily in baked goods such as french fries, potato chips, and roasted coffee.
1. History
Asparagine was first isolated in 1806 from asparagus juice, in which it is abundant – hence its name – becoming the first amino acid to be isolated. The characteristic smell observed in the urine of individuals after their consumption of asparagus is attributed to various metabolic byproducts of asparagine: in 1891, Marceli Nencki claimed that the substance responsible was methanethiol, and Robert White’s 1975 research indicated that the substances were various thioesters. Other likely possibilities include asparagine aminosuccinic monoamide. Allison and McWhirter’s 1956 research[1] indicated that some individuals do not produce this odor after asparagus consumption, and that this is autosomal; however, a re-examination in 1980 showed that these individuals are, rather, not able to detect the odor.
2. Structural function in proteins
Since the asparagine side chain can make efficient hydrogen bond interactions with the peptide backbone, asparagines are often found near the beginning and end of alpha-helices, and in turn motifs in beta sheets. Its role can be thought as “capping” the hydrogen bond interactions which would otherwise need to be satisfied by the polypeptide backbone. Glutamines have an extra methylene group, have more conformational entropy and thus are less useful in this regard.
Asparagine also provides key sites for N-linked glycosylation, modification of the protein chain with the addition of carbohydrate chains.
3. Biosynthesis
Asparagine is not an essential amino acid, which means that it can be synthesized from central metabolic pathway intermediates in humans and is not required in the diet. The precursor to asparagine is oxaloacetate. Oxaloacetate is converted to aspartate using a transaminase enzyme. The enzyme transfers the amino group from glutamate to oxaloacetate producing α-ketoglutarate and aspartate. The enzyme asparagine synthetase produces asparagine, AMP, glutamate, and pyrophosphate from aspartate, glutamine, and ATP. In the asparagine synthetase reaction, ATP is used to activate aspartate, forming β-aspartyl-AMP. Glutamine donates an ammonium group which reacts with β-aspartyl-AMP to form asparagine and free AMP.
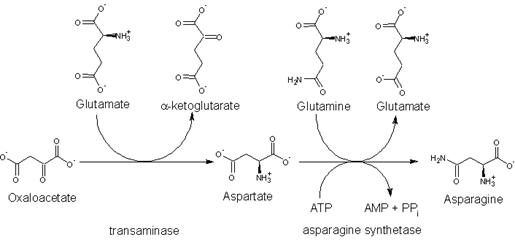
The biosynthesis of asparagine from oxaloacetate
4. Degradation
Aspartate is a glucogenic amino acid. L-asparginase hydrolyzes the amide group to form aspartate and ammonium. A transaminase converts the aspartate to oxaloacetate which can then be metabolized in the citric acid cycle or gluconeogenesis.
5. Function
The nervous system needs asparagine to maintain the equilibrium, as well as in amino acid transformation. It also plays an important role in the synthesis of ammonia.
6. Sources
Asparagus, dairy products, potatoes, beef, poultry, meat, and eggs.
7. References
1. ALLISON AC, MCWHIRTER KG (1956). “Two unifactorial characters for which man is polymorphic”. Nature 178 (4536): 748-9.
Aspartic Acid
Aspartic acid is an α-amino acid with the chemical formula HO2CCH(NH2)CH2CO2H. The L-isomer is one of the 20 proteinogenic amino acids, i.e. the building blocks of proteins. Its three letter code is asp, its one letter code is D, and its codons are GAU and GAC.[1] It is classified as an acidic amino acid, together with glutamic acid. Aspartic acid is pervasive in biosynthesis.
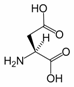
1. Role in biosynthesis of amino acids
Aspartic acid is non-essential in mammals, being produced from oxaloacetate by transamination. In plants and microorganisms, aspartic acid is the precursor to several amino acids, including four that are essential: methionine, threonine, isoleucine, and lysine. The conversion of aspartic acid to these other amino acids begins with reduction of aspartic acid to its “semialdehyde,” HO2CCH(NH2)CH2CHO.[2] Asparagine is derived from aspartic acid via transamidation:
HO2CCH(NH2)CH2CO2H + GC(O)NH2
HO2CCH(NH2)CH2CONH2 + GC(O)OH
(where GC(O)NH2 and GC(O)OH are glutamine and glutamic acid, respectively)
2. Other biochemical roles
Aspartic acid is also a metabolite in the urea cycle and participates in gluconeogenesis. It carries reducing equivalents in the malate-aspartate shuttle, which utilizes the ready interconversion of aspartate and oxaloacetate, which is the oxidized (dehydrogenated) derivative of malic acid. Aspartic acid donates one nitrogen atom in the biosynthesis of inositol, the precursor to the purine bases.
2.1. Neurotransmitter
Aspartate (the conjugate base of aspartic acid) stimulates NMDA receptors, though not as strongly as the amino acid neurotransmitter glutamate does.[3] It serves as an excitatory neurotransmitter in the brain and is an excitotoxin.
As a neurotransmitter, aspartic acid may provide resistance to fatigue and thus lead to endurance, although the evidence to support this idea is not strong.
3. References
1. IUPAC-IUBMB Joint Commission on Biochemical Nomenclature. Nomenclature and Symbolism for Amino Acids and Peptides. Recommendations on Organic & Biochemical Nomenclature, Symbols & Terminology etc.
2. Nelson, D. L.; Cox, M. M. “Lehninger, Principles of Biochemistry” 3rd Ed. Worth Publishing: New York, 2000. ISNB 1-57259-153-6.
3. Philip E. Chen, Matthew T. Geballe, Phillip J. Stansfeld, Alexander R. Johnston, Hongjie Yuan, Amanda L. Jacob, James P. Snyder, Stephen F. Traynelis, and David J. A. Wyllie. 2005. Structural Features of the Glutamate Binding Site in Recombinant NR1/NR2A N-Methyl-D-aspartate Receptors Determined by Site-Directed Mutagenesis and Molecular Modeling. Molecular Pharmacology. Volume 67, Pages 1470-1484.
4. Dunn, M. S.; Smart, B. W. “DL-Aspartic Acid” Organic Syntheses, Collected Volume 4, p.55 (1963).
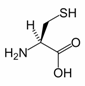
Cysteine
Cysteine is a naturally occurring, sulfur-containing amino acid that is a building block to most proteins. Cysteine is unique among the twenty common amino acids because it contains a thiol group. Thiol groups can undergo oxidation of a pair of cysteine residues is oxidised produces cystine, a disulfide-containing derivative. This reaction is reversible. The disulphide bonds of cystine are crucial to defining the structures of many proteins. Related to its redox behavoir, cysteine is incorporated into many proteins that are redox-active, such as the antioxidant glutathione. Cysteine is named after cystine, which comes from the Greek word kustis meaning bladder – cystine was first isolated from kidney stones.
Contents
1. Reactions
2. Biological functions
3. Biochemistry
4. Dietary sources
5. Production
6. Applications
7. Sheep
8. Hangover Remedy
8.1. N-acetylcysteine (NAC)
9. References
1. Reactions
Like most thiols, cystein undergoes a variety of redox reactions. Oxidation by removal of hydrogen (“dehydrogenation”) of cysteine produce the aforementioned disulfide cystine. More aggressive oxidants produce sulphfinic or sulfonic acids.
The cysteine thiol group is also nucleophilic and, thus, can undergo addition and substitution reactions. Thiol groups become much more reactive when they are ionised, and cysteine residues in proteins have pKa values close to neutrality, so are often in their reactive thiolate form in the cell.[1]
The thiol group also has a high affinity for heavy metals, so that proteins containing cysteine will bind metals such as mercury, lead and cadmium tightly.[2]
2. Biological functions
Due to this ability to undergo redox reactions, cysteine has antioxidant properties. Cysteine is an important source of sulfur in human metabolism, and although it is classified as a non-essential amino acid, cysteine may be essential for infants, the elderly, and individuals with certain metabolic disease or who suffer from malabsorption syndromes.
Cysteine is an important precursor in the production of glutathione in the body and other organisms. The systemic availability of oral glutathione (GSH) is negligible; the vast majority of it must be manufactured intracellularly. Glutathione is a tripeptide antioxidant made up of the three amino acids cysteine, glycine and glutamate. Glutamate and glycine are readily available in most North American diets, but the availability of cysteine makes it be the rate-limiting substrate for the synthesis of glutathione within the cell. It is the sulfhydryl (thiol) group (SH) of cysteine that serves as proton-donor and is responsible for the biological activity of glutathione.
The free amino acid cysteine (supplied supplementally by NAC) does not represent an ideal delivery system to the cell. Cysteine is potentially toxic and is spontaneously catabolized in the gastrointestinal tract and blood plasma. Conversely, cysteine absorbed during digestion as cystine (two cysteine molecules linked by a disulfide bond) in the gastrointestinal tract is more stable than the free amino acid cysteine. Cystine travels safely through the GI tract and blood plasma and is promptly reduced to the two cysteine molecules upon cell entry.
3. Biochemistry
Cysteine contains a thiol group, which can display nucleophilicity. Some important cysteine-derived nucleophiles include ubiquitin ligases, which transfer ubiquitin to its pendant proteins, and caspases, which engage in proteolysis in the apoptotic cycle. Inteins often function with the help of a catalytic cysteine. These roles are typically limited to the intracellular milieu, where the environment is reducing, and cysteine is not oxidized to cystine.
Cysteines play a valuable role by crosslinking proteins. This increases the molecular stability in the harsh extracellular environment, and also functions to confer proteolytic resistance (since protein export is a costly process, minimizing its necessity is advantageous). Intracellularly, disulfide bridges between cysteines within a polypeptide support the protein’s secondary structure. Insulin is an example of a protein with cystine crosslinking, where two separate peptide chains are connected by a pair of disulfide bonds.
Protein Disulfide Isomerases catalyze the proper formation of disulfide bonds; the cell transfers dehydroascorbic acid to the endoplasmic reticulum which oxidises the environment. In this environment, cysteines are generally oxidized to cystine and no longer functions as a nucleophile.
4. Dietary sources
Cysteine can be found in eggs, meat, red peppers, garlic, onions, broccoli, brussel sprouts, oats, milk, whey protein, and wheat germ. However, it is not classified as an essential amino acid, and can usually be synthesized by the human body under normal physiological conditions if a sufficient quantity of methionine is available.
5. Production
Currently the cheapest source of material from which food grade L-cysteine may be purified in high yield is by hydrolysis of molecules in human hair. Other sources include feathers and pig bristles. The companies producing cysteine by hydrolysis are located mainly in China. There is some debate whether or not consuming L-cysteine derived from human hair constitutes cannibalism. Although many other amino acids were accessible via fermentation for some years, L-Cysteine was unavailable until 2001 when a German company introduced a production route via fermentation (non-human, non-animal origin).
A source of bonded cysteine (cystine) is undenatured bovine whey protein; this is the same form as that in human breast milk.
6. Applications
Cysteine (mostly in the naturally occurring form L-cysteine) is a precursor in the food, pharmaceutical, and personal care industries. One of the largest applications is the production of flavors. For example, reacting cysteine with sugars in a Maillard reaction yields meat flavors.
L-cysteine is also used as a processing aid for baking. Small quantities (in the tens of ppm range) help to soften the dough and thus reduce processing time.
In the field of personal care, cysteine is used for permanent wave applications predominantly in Asia. Again the cysteine is used for breaking up the disulfide bonds in the hair’s keratin.
Cysteine is a very popular target for site-directed labeling experiments to investigate biomolecular structure and dynamics. Maleimides will selectively attach to cysteine using a covalent michael-addition. Site-directed spin labeling for EPR also uses cysteine extensively.
In his 2006 doctoral thesis Ville Salaspuro proposes that:
The elevated aerodigestive tract cancer risk observed in smokers and drinkers may be the result of the increased acetaldehyde exposure. Acetaldehyde produced in the oral cavity during ethanol challenge was significantly decreased by a buccal L-cysteine -releasing tablet. Also smoking-derived acetaldehyde could be totally removed by using a tablet containing L-cysteine.
In a 1994 report released by five top cigarette companies, cysteine is one of the 599 additives to cigarettes. Its use or purpose, however, is unknown, like most cigarette additives. Its inclusion in cigarettes could offer two benefits: Acting as an expectorant, since smoking increases mucus production in the lungs; and increasing the beneficial antioxidant glutathione (which is diminished in smokers).
7. Sheep
Cysteine is required by sheep in order to produce wool, however it is an essential amino-acid that cannot be synthesised by the sheep and must be taken in as food from grass. This means that during drought conditions sheep stop producing wool; however, transgenic sheep have been developed which can make their own cysteine. A little concentration of cysteine is used to break the disulfide bonds during the solublisation of recombinant protein preparation.
8. Hangover Remedy
Cysteine has been linked to aiding in the remedy of certain hangover symptoms. It directly counteracts the poisonous effects of acetaldehyde, a particularly toxic by-product of alcohol in the human body. Cysteine attracts the toxin, breaking it down into the non-toxic acetate, a substance similar to vinegar. The actual effectiveness of consuming cysteine as part of a hangover remedy is unclear.
8.1. N-acetylcysteine (NAC)
N-acetyl-L-cysteine (NAC) is a derivatives of cysteine wherein an acetyl group is attached to the nitrogen atom. This compound is sometimes considered as a dietary supplement. NAC is often used as a cough medicine as it breaks up the disulfide bonds in the mucus and thus liquefies it, making it easier to cough up. NAC is also used as a dietary supplement as already indicated above, as well as a specific antidote in cases of acetominophen overdose.
9. References
1. Bulaj G, Kortemme T, Goldenberg D (1998). “Ionization-reactivity relationships for cysteine thiols in polypeptides.”. Biochemistry 37 (25): 8965-72. PMID 9636038.
2. Baker D, Czarnecki-Maulden G (1987). “Pharmacologic role of cysteine in ameliorating or exacerbating mineral toxicities.”. J Nutr 117 (6): 1003-10. PMID 3298579.
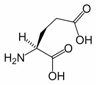
Glutamic Acid
Glutamic acid (Glu, E), also referred to as glutamate (the anion), is one of the 20 proteinogenic amino acids. It is not among the essential amino acids.
Contents
1. Structure
2. Synthesis
2.1. Natural
3. Function
3.1. In metabolism
3.2. As a neurotransmitter
3.3. In brain nonsynaptic glutamatergic signaling circuits
3.3.1. GABA precursor
4. Sources and absorption
5. Pharmacology
6. References
1. Structure
As its name indicates, it is acidic, with a carboxylic acid component to its side chain. Generally either the amino group will be protonated or one or both of the carboxylic groups will be deprotonated. At neutral pH all three groups are ionized and the species has a charge of -1. The pKa value for Glutamic acid is 4.1. This means that at pH below this value it will be protonated (COOH) and at pH above this value it will be deprotonated (COO-)
2. Synthesis
2.1. Natural
Reactants | Products | Enzymes |
Glutamine + H2O | → Glu + NH3 | GLS, GLS2 |
NAcGlu + H2O | → Glu + Acetate | (unknown) |
α-ketoglutarate + NADPH + NH4+ | → Glu + NADP+ + H2O | GLUD1, GLUD2 |
α-ketoglutarate + α-amino acid | → Glu + α-oxo acid | transaminase |
1-pyrroline-5-carboxylate + NAD+ + H2O | → Glu + NADH | ALDH4A1 |
N-formimino-L-glutamate + FH4 | ⇌ Glu + 5-formimino-FH4 | FTCD |
3. Function
3.1. In metabolism
Glutamate is a key molecule in cellular metabolism. In humans, dietary proteins are broken down by digestion into amino acids, which serves as metabolic fuel for other functional roles in the body. A key process in amino acid degradation is transamination, in which the amino group of an amino acid is transferred to an α-ketoacid, typically catalysed by a transaminase. The reaction can be generalised as such:
R1-amino acid + R2-α-ketoacid ⇌ R1-α-ketoacid + R2-amino acid
A very common α-ketoacid is α-ketoglutarate, an intermediate in the citric acid cycle. When α-ketoglutarate undergoes transamination, it always results in glutamate being formed as the corresponding amino acid product. The resulting α-ketoacid product is often a useful one as well, which can contribute as fuel or as a substrate for further metabolism processes. Examples are as follows:
alanine + α-ketoglutarate ⇌ pyruvate + glutamate
aspartate + α-ketoglutarate ⇌ oxaloacetate + glutamate
Both pyruvate and oxaloacetate are key components of cellular metabolism, contributing as substrates or intermediates in fundamental processes such as glycolysis, gluconeogenesis and also the citric acid cycle.
Glutamate also plays an important role in the body’s disposal of excess or waste nitrogen. Glutamate undergoes deamination, an oxidative reaction catalysed by glutamate dehydrogenase, as follows:
glutamate + water + NAD+ → α-ketoglutarate + NADH + ammonia + H+
Ammonia (as ammonium) is then excreted predominantly as urea, synthesised in the liver. Transamination can thus be linked to deamination, effectively allowing nitrogen from the amine groups of amino acids to be removed, via glutamate as an intermediate, and finally excreted from the body in the form of urea.
3.2. As a neurotransmitter
Glutamate is the most abundant fast excitatory neurotransmitter in the mammalian nervous system. At chemical synapses, glutamate is stored in vesicles. Nerve impulses trigger release of glutamate from the pre-synaptic cell. In the opposing post-synaptic cell, glutamate receptors, such as the NMDA receptor, bind glutamate and are activated. Because of its role in synaptic plasticity, it is believed that glutamic acid is involved in cognitive functions like learning and memory in the brain.
Glutamate transporters[3] are found in neuronal and glial membranes. They rapidly remove glutamate from the extracellular space. In brain injury or disease, they can work in reverse and excess glutamate can accumulate outside cells. This process causes calcium ions to enter cells via NMDA receptor channels, leading to neuronal damage and eventual cell death, and is called excitotoxicity. The mechanisms of cell death include:
• Damage to mitochondria from excessively high intracellular Ca2+[4].
• Glu/Ca2+-mediated promotion of transcription factors for pro-apoptotic genes, or downregulation of transcription factors for anti-apoptotic genes.
Excitotoxicity due to glutamate occurs as part of the ischemic cascade and is associated with stroke and diseases like amyotrophic lateral sclerosis, lathyrism, and Alzheimer’s disease.
Glutamic acid has been implicated in epileptic seizures. Microinjection of glutamic acid into neurons produces spontaneous depolarisations around one second apart, and this firing pattern is similar to what is known as paroxysmal depolarizing shift in epileptic attacks. This change in the resting membrane potential at seizure foci could cause spontaneous opening of voltage activated calcium channels, leading to glutamic acid release and further depolarization.
Experimental techniques to detect glutamate in intact cells include using a genetically-engineered nanosensor[2]. The sensor is a fusion of a glutamate-binding protein and two fluorescent proteins. When glutamate binds, the fluorescence of the sensor under ultraviolet light changes by resonance between the two fluorophores. Introduction of the nanosensor into cells enables optical detection of the glutamate concentration. Synthetic analogs of glutamic acid that can be activated by ultraviolet light have also been described[6]. This method of rapidly uncaging by photostimulation is useful for mapping the connections between neurons, and understanding synapse function.
3.3. In brain nonsynaptic glutamatergic signaling circuits
Extracellular glutamate in Drosophila brains has been found to regulate postsynaptic glutamate receptor clustering, via a process involving receptor desensitization[7]. A gene expressed in glial cells actively transports glutamate into the extracellular space[7], while in the nucleus accumbens stimulating group II metabotropic glutamate receptors was found to reduce extracellular glutamate levels[8]. This raises the possibility that this extracellular glutamate plays an “endocrine-like” role as part of a larger homeostatic system.
3.3.1. GABA precursor
Glu also serves as the precursor for the synthesis of the inhibitory GABA in GABA-ergic neurons. This reaction is catalyzed by GAD, glutamic acid decarboxylase, which is most abundant in cerebellum and pancreas.
Stiff-man syndrome is a neurologic disorder caused by anti-GAD antibodies, leading to a decrease in GABA synthesis and therefore, impaired motor function such as muscle stiffness and spasm. Since the pancreas is also abundant for the enzyme GAD, a direct immunological destruction occurs in the pancreas and the patients will have diabetes mellitus.
4. Sources and absorption
Glutamic acid is present in a wide variety of foods and is responsible for one of the five basic tastes of the human sense of taste (umami), especially in its physiological form, the sodium salt of glutamate in a neutral pH. Ninety-five percent of the dietary glutamate is metabolized by intestinal cells in a first pass [5].
Overall, glutamic acid is the single largest contributor to intestinal energy. As a source for umami, the sodium salt of glutamic acid, monosodium glutamate (MSG) is used as a food additive to enhance the flavor of foods, although an identical effect can be achieved by mixing and cooking together different ingredients rich in this amino acid and other umami substances as well.
Another source of MSG is fruits, vegetables and nuts that have been sprayed with Auxigro. Auxigro is a growth enhancer that contains 30% glutamic acid.
China-based Fufeng Group Limited is the largest producer of Glutamic Acid in the world, with capacity increasing to 300,000 tons at the end of 2006 from 180,000 tons during 2006, putting them at 25-30% of the chinese market. Meihua is the second largest Chinese producer. Together, the top five producers have roughly 50% share in China. Chinese demand is roughly 1.1 million tons per year, while global demand, including China, is 1.7 million tons per year.
5. Pharmacology
The drug phencyclidine (more commonly known as PCP) antagonizes glutamic acid non-competitively at the NMDA receptor. For the same reasons, sub-anaesthetic doses of Ketamine have strong dissociative and hallucinogenic effects. Glutamate does not easily pass the blood brain barrier, but: “glutamate flux from plasma into brain is mediated by a high affinity transport system at the Blood-Brain Barrier” [1]. It can also be converted into glutamine.
Glutamate transport and supply are obvious targets for the treatment of epilepsy, therefore. In particular Glutamate Restriction Diets are now claiming success anecdotally, by limiting or eliminating intake of wheat, peanut, soy and bean. No similar diets for schizophrenia are known.
6. References
1. Nelson DL and Cox MM. Lehninger Principles of Biochemistry, 4th edition.
2. Okumoto, S., et al. (2005). “Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors”. Proceedings of the National Academy of Sciences U.S.A 102 (24): 8740-8745.
3. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Brain Res Rev. 2004 Jul; 45(3):250-65.
4. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol. 1989 Jul;36(1):106-12.
5. Reeds, P.J., et al. (2000). “Intestinal glutamate metabolism”. Journal of Nutrition 130 (4s): 978S-982S.
6. Corrie, J.E., et al. (1993). “Postsynaptic activation at the squid giant synapse by photolytic release of L-glutamate from a ‘caged’ L-glutamate”. Journal of Physiology 465 (Jun): 1-8.
7. Augustin H, Grosjean Y, Chen K, Sheng Q, Featherstone DE (2007). “Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo”. Journal of Neuroscience 27 (1): 111-123.
8. Zheng Xi, Baker DA, Shen H, Carson DS, Kalivas PW (2002). “Group II metabotropic glutamate receptors modulate extracellular glutamate in the nucleus accumbens”. Journal of Pharmacology and Experimental Therapeutics 300 (1): 162-171.
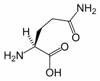
Glutamine
Glutamine (Gln, Q) is one of the 20 amino acids encoded by the standard genetic code. Its side chain is an amide; it is formed by replacing a side-chain hydroxyl of glutamic acid with an amine functional group.
1. Biochemistry
1.1. Formation and Nomenclature
Glutamine is genetically coded for by the RNA codons CAA and CAG. Glutamine’s three-letter abbreviation is Gln, and its one-letter abbreviation is Q. A three-letter designation for either glutamine or glutamic acid is Glx (one-letter abbreviation: Z).
Like other amino acids, glutamine is biochemically important as a constituent of proteins. Glutamine is also crucial in nitrogen metabolism. Ammonia (formed by nitrogen fixation) is assimilated into organic compounds by converting glutamic acid to glutamine. The enzyme that accomplishes this is called glutamine synthetase. Glutamine can, hence, be used as a nitrogen donor in the biosynthesis of many compounds, including other amino acids, purines, and pyrimidines.
Artificial glutamine synthesis was first reported in 1933 in the Bergmann-Zervas carbobenzoxy method [1]
2. Occurrences in Nature
Glutamine is found in foods high in proteins, such as fish, red meat, beans, and dairy products.
3. References
1. Bergmann, M., Zervas, L., and Salzmann, L., Ber. them. Ges., 99, 1233 (1933).
Glycine
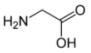
Glycine is the organic compound with the formula HO2CCH2NH2. It is one of the 20 amino acids commonly found in animal proteins. Its three letter code is gly, its one letter code is G, and its codons are GGU, GGC, GGA and GGG.[1] Because of its structural simplicity, this compact amino acid tends to be evolutionarily conserved in, for example, cytochrome c, myoglobin, and hemoglobin. Glycine is the unique amino acid that is not optically active. Most proteins contain only small quantities of glycine. A notable exception is collagen, which contains about one-third glycine.
1. Biosynthesis
Glycine is not essential to the human diet, since it is synthesized in the body. It is biosynthesized from the amino acid serine. The enzyme serine hydroxymethyl transferase catalyses this transformation:[2]
HO2CCH(NH2)CH2OH + H2folate → HO2CCH2NH2 + CH2-folate + H2O
2. Physiological function
2.1. As a biosynthetic intermediate
Glycine is a building block to numerous species. Aminolevulinic acid, the key precursor to porphyrins is biosynthesized from glycine and succinoyl coenzyme A. Glycine provides the central C2N subunit of all purines.[2]
2.2. As a neurotransmitter
Glycine is an inhibitory neurotransmitter in the central nervous system, especially in the spinal cord, brainstem, and retina. When glycine receptors are activated, chloride enters the neuron via ionotropic receptors, causing an Inhibitory postsynaptic potential (IPSP). Strychnine is an antagonist at ionotropic glycine receptors. Glycine is a required co-agonist along with glutamate for NMDA receptors. In contrast to the inhibitory role of glycine in the spinal cord, this behaviour is facilitated at the (NMDA) glutaminergic receptors which are excitatory. The LD50 of glycine is 7930 mg/kg in rats (oral),[3] and it usually causes death by hyperexcitability.
3. Presence in the interstellar medium
In 1994 a team of astronomers at the University of Illinois, led by Lewis Snyder, claimed that they had found the glycine molecule in space. It turned out that, with further analysis, this claim could not be confirmed. Nine years later, in 2003, Yi-Jehng Kuan from National Taiwan Normal University and Steve Charnley claimed that they detected interstellar glycine toward three sources in the interstellar medium [1]. They claimed to have identified 27 spectral lines of glycine utilizing a radio telescope. According to computer simulations and lab-based experiments, glycine was probably formed when ices containing simple organic molecules were exposed to ultraviolet light [4].
In October 2004, Snyder and collaborators reinvestigated the glycine claim in Kuan et al. (2003). In a rigorous attempt to confirm the detection, Snyder showed that glycine was not detected in any of the three claimed sources [2].
Should the glycine claim be substantiated, it does not prove that life exists outside the Earth, but certainly makes that possibility more plausible by showing that amino acids can be formed in the interstellar medium. The finding would also indirectly support the idea of panspermia, the theory that life was brought to Earth from space. As in Miller-Urey experiment, it should be noted that glycine is but the simplest of amino acids.
4. References
1. Kuan YJ, Charnley SB, Huang HC, et al. (2003) Interstellar glycine. ASTROPHYS J 593 (2): 848-867
2. Snyder LE, Lovas FJ, Hollis JM, et al. (2005) A rigorous attempt to verify interstellar glycine. ASTROPHYS J 619 (2): 914-930
3. The Physical and Theoretical Chemistry Laboratory Oxford University (2005). Retrieved on 2006-11-01.
4. Dawson, R.M.C., Elliott, D.C., Elliott, W.H., and Jones, K.M., Data for Biochemical Research (3rd edition), pp. 1-31 (1986)
Histidine
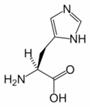
Histidine (His, H) is one of the 20 most common natural amino acids present in proteins. In the nutritional sense, in humans, histidine is considered an essential amino acid, but mostly only in children.
1. Chemical properties
The imidazole side chains and the relatively neutral pKa of histidine (ca 6.0) mean that relatively small shifts in cellular pH will change its charge. For this reason, this amino acid side chain finds its way into considerable use as a co-ordinating ligand in metalloproteins, and also as a catalytic site in certain enzymes. The imidazole side chain has two nitrogens with different properties: One is bound to hydrogen and donates its lone pair to the aromatic ring and as such is slighty acidic, whereas the other one donates only one electron pair to the ring so it has a free lone pair and is basic. These properties are exploited in different ways in proteins. In catalytic triads, the basic nitrogen of histidine is used to abstract a proton from serine, threonine or cysteine to activate it as a nucleophile. In a histidine proton shuttle, histidine is used to quickly shuttle protons, it can do this by abstracting a proton with its basic nitrogen to make a positively-charged intermediate and then use another molecule, a buffer, to extract the proton from its acidic nitrogen. In carbonic anhydrases, a histidine proton shuttle is utilized to rapidly shuttle protons away from a zinc-bound water molecule to quickly regenerate the active form of the enzyme.
2. Metabolism
The amino acid is a precursor for histamine and carnosine biosynthesis.

Conversion of histidine to histamine by histidine decarboxylase
The enzyme histidine ammonia-lyase converts histidine into ammonia and urocanic acid. A deficiency in this enzyme is present in the rare metabolic disorder histidinemia.
3. Sources
Histidine is found in fruits such as bananas and grapes, meat and poultry, and milk and milk products. It is also found in root vegetables and all green vegetables, though in lesser quantities.
4. Forms
There are two enantiomers: D-histidine and L-histidine.
5. History
Histidine was first isolated in 1896 by German physician Albrecht Kossel.
Isoleucine
Isoleucine is an α-amino acid with the chemical formula HO2CCH(NH2)CH(CH3)CH2CH3. Its three letter code is ILE and its one letter code is I. It is an essential amino acid, which means that humans cannot synthesise it, so it must be part of our diet. With a hydrocarbon side chain, Isoleucine is classified as a hydrophobic amino acid.
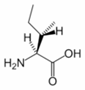
Together with threonine, isoleucine is one of two common amino acids that has a chiral side chain. Four stereoisomers of isoleucine are possible, including two possible diastereomers of L-isoleucine. However, isoleucine present in nature exists in one enantiomeric form, (2S,3S)-2-amino-3-methylpentanoic acid.
1. Biosynthesis
As an essential amino acid, isoleucine is not synthesized in animals, hence it must be ingested, usually as a component of proteins. In plants and microorganisms, it is synthesized via several steps starting from pyruvic acid and alpha-ketoglutarate. Enzymes involved in this biosynthesis include:[1]
• acetolactate synthase
• acetohydroxy acid isomeroreductase
• dihydroxyacid dehydratase
• valine aminotransferase
2. Isomers of isoleucine
Forms of Isoleucine |
|||||||
Common name: |
isoleucine |
D-isoleucine |
L-isoleucine |
DL-isoleucine |
allo-D-isoleucine |
allo-L-isoleucine |
allo-DL-isoleucine |
Synonyms: |
|
(R)-Isoleucine |
L(+)-Isoleucine |
(R*,R*)-isoleucine |
|
alloisoleucine |
|
PubChem: |
CID 791 |
CID 94206 |
CID 6306 |
CID 76551 |
|
|
|
EINECS number: |
207-139-8 |
206-269-2 |
200-798-2 |
|
216-143-9 |
216-142-3 |
221-464-2 |
CAS number: |
443-79-8 |
319-78-8 |
73-32-5 |
|
1509-35-9 |
1509-34-8 |
3107-04-8 |
3. Synthesis
Isoleucine can be synthesized in a multistep procedure starting from 2-bromobutane and diethylmalonate.[2] Synthetic isoleucine was originally reported in 1905.[3]
4. Dietary aspects
Rich sources of isoleucine are eggs, chicken, pork, mutton, pulses, soya beans, cottage cheese, milk, piyal seeds, cashew nuts, and cereal grains.
5. References
1. Nelson, D. L.; Cox, M. M. “Lehninger, Principles of Biochemistry” 3rd Ed. Worth Publishing: New York, 2000.
2. Marvel, C. S. “dl-Isoleucine” Organic Syntheses, Collected Volume 3, p.495 (1955).
3. Bouveault and Locquin, Compt. rend., 141, 115 (1905).
Leucine
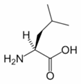
Leucine is an α-amino acid with the chemical formula HO2CCH(NH2)CH2CH(CH3)2. Its three letter code is leu, its one letter code is L, and its codons are UUA, UUG, CUU, and CUC. It is an essential amino acid, which means that humans cannot synthesise it. With a hydrocarbon side chain, leucine is classified as a hydrophobic amino acid. It is an isomer of isoleucine.
1. Biosynthesis
As an essential amino acid, leucine is not synthesized in animals, hence it must be ingested, usually as a component of proteins. It is synthesized in plants and microorganisms via several steps starting from pyruvic acid. The initial part of the pathway also leads to valine. The intermediate α-ketovalerate is converted to α-isopropylmalate and then β-isopropylmalate, which is dehydrogenated to α-ketoisocaproate, which in the final step undergoes reductive amination. Enzymes involved in a typical biosynthesis include:[1]
• acetolactate synthase
• acetohydroxy acid isomeroreductase
• dihydroxyacid dehydratase
• α-isopropylmalate synthase
• α-isopropylmalate isomerase
• leucine aminotransferase
2. Dietary aspects
Major food sources of leucine include whole grains, milk products, eggs (~1 g/100g), pork, beef, chicken, pulses (such as soybeans (~3 g/100g), chick peas and lentils) and leaf vegetables.
3. References
1. Nelson, D. L.; Cox, M. M. “Lehninger, Principles of Biochemistry” 3rd Ed. Worth Publishing: New York, 2000.
Lysine
Lysine is an α-amino acid with the chemical formula HO2CCH(NH2)(CH2)4NH2. Its three letter code is Lys, its one letter code is K, and its codons are AAA and AAG. This amino acid is essential amino acid, which means that humans cannot synthesise it. Lysine is a basic, as are arginine and histidine. The δ-amino group often participates in hydrogen bonding and as a general base in catalysis.
Common posttranslational modifications include methylation of the e-amino group, giving methyl-, dimethyl-, and trimethyllysine. The latter occurs in calmodulin. Other posttranslational modifications include acetylation. Collagen contains hydroxylysine which is derived from lysine by lysyl hydroxylase. O-Glycosylation of lysine residues in the endoplasmic reticulum or Golgi apparatus is used to mark certain proteins for secretion from the cell.
1. Biosynthesis
As an essential amino acid, lysine is not synthesized in animals, hence it must be ingested as lysine or lysine-containing proteins. In plants and microorganisms, it is synthesized from aspartic acid, which is first converted to β-aspartyl-semialdehyde. Cyclization gives dihydropicolinate, which is reduced to Δ1-piperidine-2,6-dicarboxylate. Ring-opening of this heterocycle gives a series of derivatives of pimelic acid, ultimately affording lysine. Enzymes involves in this biosynthesis include:[1]
• aspartokinase
• β-aspartate semialdehyde dehydrogenase
• dihydropicolinate synthase
• Δ1-piperdine-2,6-dicarboxylate dehydrogenase
• N-succinyl-2-amino-6ketopimelate synthase
• succinyl diaminopimelate aminotransferase
• succinyl diaminopimelate desuccinylase
• diaminopimelate epimerase
• diaminopimelate decarboxylase
2. Metabolism
Lysine is metabolised in mammals to give acetyl-CoA, via an initial transamination with α-ketoglutarate. The bacterial degradation of lysine yields cadaverine by decarboxylation.
3. Synthesis
Synthetic, racemic lysine has long been known.[2] A practical synthesis starts from caprolactam.[3]
4. Dietary sources
The human nutritional requirement is 1–1.5 g daily. Used as a dietary supplement. It is the limiting amino acid in all cereal grains, but is plentiful in all pulses (legumes). Fish are also quite rich in lysine. Plants that contain significant amounts of lysine include:[citation needed]
• Buffalo Gourd (10,130–33,000 ppm) in seed
• Berro, Watercress (1,340–26,800 ppm) in herb.
• Soybean (24,290–26,560 ppm) in seed.
• Carob, Locust Bean, St.John’s-Bread (26,320 ppm) in seed;
• Common Bean (Black Bean, Dwarf Bean, Field Bean, Flageolet Bean, French Bean, Garden Bean, Green Bean, Haricot, Haricot Bean, Haricot Vert, Kidney Bean, Navy Bean, Pop Bean, Popping Bean, Snap Bean, String Bean, Wax Bean) (2,390–25,700 ppm) in sprout seedling;
• Ben Nut, Benzolive Tree, Jacinto (Sp.), Moringa (aka Drumstick Tree, Horseradish Tree, Ben Oil Tree), West Indian Ben (5,370–25,165 ppm) in shoot.
• Lentil (7,120–23,735 ppm) in sprout seedling.
• Asparagus Pea, Winged Bean (aka Goa Bean) (21,360–23,304 ppm) in seed.
• Fat Hen (3,540–22,550 ppm) in seed.
• Lentil (19,570–22,035 ppm) in seed.
• White Lupin (19,330–21,585 ppm) in seed.
• Black Caraway, Black Cumin, Fennel-Flower, Nutmeg-Flower, Roman Coriander (16,200–20,700 ppm) in seed.
• Spinach (1,740–20,664 ppm).
• Amaranth, Quinoa
5. Properties
L-Lysine is a necessary building block for all protein in the body. L-Lysine plays a major role in calcium absorption; building muscle protein; recovering from surgery or sports injuries; and the body’s production of enzymes, and antibodies.
6. Clinical significance
It has been suggested that lysine may be beneficial for those with herpes simplex infections.[4] However, more research is needed to fully substantiate this claim. For more information, refer to Herpes simplex – Lysine.
7. References
1. Nelson, D. L.; Cox, M. M. “Lehninger, Principles of Biochemistry” 3rd Ed. Worth Publishing: New York, 2000. ISBN 1-57259-153-6.
2. Braun, J. V. “Synthese des inaktiven Lysins aus Piperidin” Berichte der deutschen chemischen Gesellschaft 1909, Volume 42, p 839-846.
3. Eck, J. C.; Marvel, C. S. “dl-Lysine Hydrochlorides” Organic Syntheses, Collected Volume 2, p.374 (1943).
4. Griffith RS, Norins AL, Kagan C. (1978). “A multicentered study of lysine therapy in Herpes simplex infection”. Dermatologica. 156 (5): 257-267.
Methionine
Methionine is an α-amino acid with the chemical formula HO2CCH(NH2)CH2CH2SCH3. This essential is classified as nonpolar. Together with cysteine, methionine is one of two sulfur-containing proteinogenic amino acids. Its derivative S-adenosyl methionine (SAM) serves as a methyl donor. Methionine plays a role in the biosynthesis of cysteine, carnitine, and taurine (by the transsulfuration pathway), lecithin production, the synthesis of phosphatidylcholine, and other phospholipids. Improper conversion of methionine can lead to atherosclerosis.
Methionine is one of only two amino acids encoded by a single codon (AUG) in the standard genetic code (tryptophan, encoded by UGG, is the other). The codon AUG is also significant, in that it carries the “Start” message for a ribosome to begin protein translation from mRNA. As a consequence, methionine is incorporated into the N-terminal position of all proteins in eukaryotes and archaea during translation, although it is usually removed by post-translational modification.
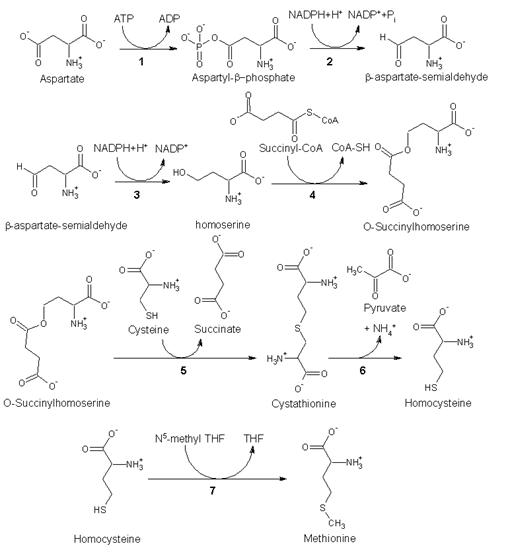
1. Biosynthesis
As an essential amino acid, methionine is not synthesized in humans, hence we must ingest methionine or methionine-containing proteins. In plants and microorganisms, methionine is synthesized via a pathway that uses both aspartic acid and cysteine. First, aspartic acid is converted via β-aspartyl-semialdehyde into homoserine, introducing the pair of contiguous methylene groups. Homoserine converts to O-succinyl homoserine, which then reacts with cysteine to produce cystathionine, which is cleaved to yield homocysteine. Subsequent methylation of the thiol group by folates affords methionine. Both cystathionine-γ-synthase and cystathionine-β-lyase require Pyridoxyl-5′-phosphate as a cofactor, whereas homocysteine methyltransferase requires Vitamin B12 as a cofactor.[1]
Enzymes involved in methionine biosynthesis:
• aspartokinase
• β-aspartate semialdehyde dehydrogenase
• homoserine dehydrogenase
• homoserine acyltransferase
• cystathionine-γ-synthase
• cystathionine-β-lyase
• methionine synthase (in mammals, this step is performed by homocysteine methyltransferase)
2. Other biochemical pathways
Although mammals cannot synthesize methionine, they can still utilize it in a variety of biochemical pathways:
Methionine is converted to S-adenosylmethionine (SAM) by (1) methionine adenosyltransferase. SAM serves as a methyl-donor in many (2) methyltransferase reactions and is converted to S-adenosylhomocysteine (SAH). (3) adenosylhomocysteinase converts SAH to homocysteine.
There are two fates of homocysteine.
• First, methionine can be regenerated from homocysteine via (4) methionine synthase. It can also be remethylated using glycine betaine (NNN-trimethyl glycine) to methionine via the enzyme Betaine-homocysteine methyltransferase (E.C.2.1.1.5, BHMT). BHMT makes up to 1.5% of all the soluble protein of the liver, and recent evidence suggests that it may have a greater influence on methionine and homocysteine homeostasis than Methionine sythase.
• Alternatively, homocysteine can be converted to cysteine. (5) cystathionine-β-synthase (a PLP-dependent enzyme) combines homocysteine and serine to produce cystathionine. Instead of degrading cystathionine via cystathionine-β-lyase as in the biosynthetic pathway, cystathionine is broken down to cysteine and α-ketobutyrate via (6) cystathionine-γ-lyase. (7) α-ketoacid dehydrogenase converts α-ketobutyrate to propionyl-CoA, which is metabolized to succinyl-CoA in a three-step process (see propionyl-CoA for pathway).
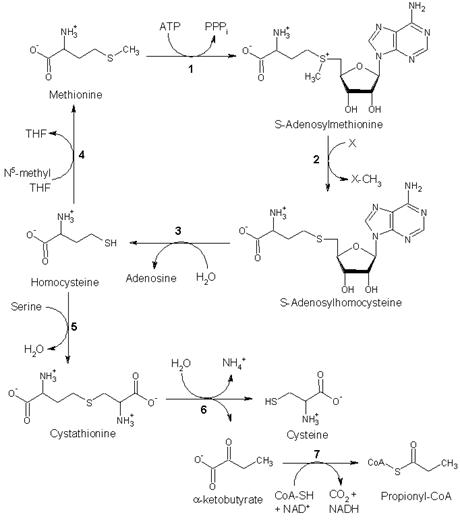
Fates of methionine
3. Synthesis
Racemic methionine can be synthesized from diethyl sodium phthalimidomalonate, (C6H4(CO)2NC(CO2Et)2), by alkylation with chloroethylmethylsulfide, ClCH2CH2SCH3 followed by hydrolysis and decarboxylation.[2]
4. Dietary aspects
High levels of methionine can be found in sesame seeds, Brazil nuts, fish, meat, and some seeds. Most fruit and vegetables contain very little, although peppers and spinach are the best sources.
5. References
1. Nelson, D. L.; Cox, M. M. “Lehninger, Principles of Biochemistry” 3rd Ed. Worth Publishing: New York, 2000. ISBN 1-57259-153-6.
2. Barger, G.; Weichselbaum, T. E. “dl-Methionine” Organic Syntheses, Collected Volume 2, p.384 (1943).
Phenylanine
Phenyl alanine is an α-amino acid with the formula HO2CCH(NH2)CH2C6H5. This essential amino acid is classified as nonpolar because of the hydrophobic nature of the benyl side chain. The codons for L-phenylalanine are UUU and UUC. It is a white, powdery solid. L-Phenylalanine (LPA) is an electrically-neutral amino acid, one of the twenty common amino acids used to biochemically form proteins, coded for by DNA.
Contents
1. Biosynthesis
2. Other biological roles
2.1. Phenylketonuria
3. Dietary aspects
4. D- and DL-phenylalanine
5. History
6. References
1. Biosynthesis
Phenylalanine cannot be made by animals, which have to obtain it from their diet. It is produced by plants and most microorganisms from prephenate, an intermediate on the shikimate pathway.[1]
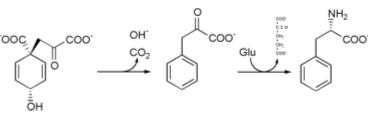
Prephenate is decarboxylated with loss of the hydroxyl group to give phenylpyruvate. This species is transaminated using glutamate as the nitrogen source to give phenylalanine and α-ketoglutarate.
2. Other biological roles
L-phenylalanine can also be converted into L-tyrosine, another one of the DNA-encoded amino acids. L-tyrosine in turn is converted into L-DOPA, which is further converted into dopamine, norepinephrine (noradrenaline), and epinephrine (adrenaline) (the latter three are known as the catecholamines).
Phenylalanine uses the same active transport channel as tryptophan to cross the blood-brain barrier, and, in large quantities, interferes with the production of serotonin.
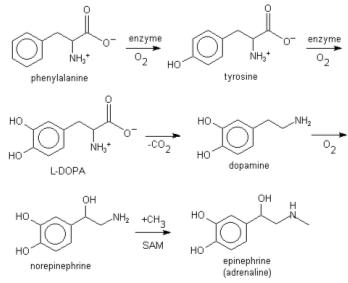
Lignin is derived from phenylalanine and from tyrosine. Phenylalanine is converted to cinnamic acid by the enzyme phenylalanine ammonia lyase.[1]
2.1. Phenylketonuria
The genetic disorder phenylketonuria (PKU) is the inability to metabolize phenylalanine. Individuals with this disorder are known as “phenylketonurics” and must abstain from consumption of phenylalanine. This dietary restriction also applies to pregnant women with hyperphenylalanine (high levels of phenylalanine in blood) because they do not properly metabolize the amino acid phenylalanine. Phenylalanine is present in many sugarless gums, Monster Munch crisps, sugarless soft drinks (such as Diet Coke, and Diet Pepsi), some forms of Lipton Tea, Icebreakers Mints, Clear Splash flavored water, and a number of other food products, all of which must be labeled: “Phenylketonurics: Contains phenylalanine.” Phenylalanine itself is not present in the food. Rather, the artificial sweetener aspartame sold under the names “Equal” and “NutraSweet” contain Aspartame, an ester that is hydrolyzed in the body to give phenylalanine, aspartic acid, and methanol (wood alcohol). Thus, aspartame should is problematic for persons with PKU. The amounts produced by aspartame pose a risk however, as far larger quantities of the amino acid would be obtained through consuming normal protein. Interestingly, the macaque genome was recently sequenced and it was found that macaques naturally have a mutation that is found in humans who have PKU.[1]
3. Dietary aspects
Phenylalanine is contained in most protein-rich foods. Especially good sources are dairy products (curd, milk, cottage cheese), avocados, pulses and legumes (particularly peanuts and lima beans), nuts (pistachios, almonds), seeds (piyal seeds), leafy vegetables, whole grains, poultry, fish, other seafoods, and some diet beverages.
4. D- and DL-phenylalanine
D-phenylalanine (DPA) either as a single enantiomer or as a component of the racemic mixture is available through conventional organic synthesis. It does not participate in protein biosynthesis although it is found in proteins, in small amounts, particularly aged proteins and food proteins that have been processed. The biological functions of D-amino acids remain unclear. Some D-amino acids, such as D-phenylalanine, may have pharmacologic activity.
DL-Phenylalanine is marketed as a nutritional supplement for its putative analgesic and antidepressant activities. The putative analgesic activity of DL-phenylalanine may be explained by the possible blockage by D-phenylalanine of enkephalin degradation by the enzyme carboxypeptidase A. The mechanism of DL-phenylalanine’s putative antidepressant activity may be accounted for by the precursor role of L-phenylalanine in the synthesis of the neurotransmitters norepinephrine and dopamine. Elevated brain norepinephrine and dopamine levels are thought to be associated with antidepressant effects.
D-phenylalanine is absorbed from the small intestine, following ingestion, and transported to the liver via the portal circulation. A fraction of D-phenylalanine appears to be converted to L-phenylalanine. D-phenylalanine is distributed to the various tissues of the body via the systemic circulation. D-phenylalanine appears to cross the blood-brain barrier with less efficiency than L-phenylalanine. A fraction of an ingested dose of D-phenylalanine is excreted in the urine.
5. History
The genetic codon for phenylalanine was the first to be discovered. Marshall W. Nirenberg discovered that insertion of m-RNA made up of multiple uracil repeats into E. coli, the bacterium produced a new protein, made up solely of repeated phenylalanine amino acids.
6. References
1. Nelson, D. L.; Cox, M. M. “Lehninger, Principles of Biochemistry” 3rd Ed. Worth Publishing: New York, 2000. ISBN 1-57259-153-6.
Proline
L-Proline is one of the twenty proteinogenic units which are used in living organisms as the building blocks of proteins. The other nineteen units are all primary amino acids, but due to the cyclic binding of the three-carbon side chain to the nitrogen of the backbone, proline lacks a primary amine group (−NH2). The nitrogen in proline is properly referred to as a secondary amine. Proline is sometimes called an imino acid, although the IUPAC definition of an imine requires a carbon-nitrogen double bond. In biological terminology, however, the category “amino acids” is generally taken to include proline.
1. Structural properties
The distinctive cyclic structure of proline’s side chain locks its φ backbone dihedral angle at approximately -75°, giving proline an exceptional conformational rigidity compared to other amino acids. Hence, proline loses less conformational entropy upon folding, which may account for its higher prevalence in the proteins of thermophilic organisms. Proline acts as a structural disruptor in the middle of regular secondary structure elements such as alpha helices and beta sheets; however, proline is commonly found as the first residue of an alpha helix and also in the edge strands of beta sheets. Proline is also commonly found in turns, which may account for the curious fact that proline is usually solvent-exposed, despite having a completely aliphatic side chain. Because proline lacks a hydrogen on the amide group, it cannot act as a hydrogen bond donor, only as a hydrogen bond acceptor.
Multiple prolines and/or hydroxyprolines in a row can create a polyproline helix, the predominant secondary structure in collagen. The hydroxylation of proline by prolyl hydroxylase (or other additions of electron-withdrawing substituents such as fluorine) increases the conformational stability of collagen significantly. Hence, the hydroxylation of proline is a critical biochemical process for maintaining the connective tissue of higher organisms. Severe diseases such as scurvy can result from defects in this hydroxylation, e.g., mutations in the enzyme prolyl hydroxylase or lack of the necessary ascorbate (vitamin C) cofactor.
Sequences of proline and 2-aminoisobutyric acid (Aib) also form a helical turn structure.
2. Cis-trans isomerization
Peptide bonds to proline and other N-substituted amino acids (such as sarcosine) are able to populate both the cis and trans isomers. Most peptide bonds prefer overwhelmingly to adopt the trans isomer (typically 99.9% under unstrained conditions), chiefly because the amide hydrogen (trans isomer) offers less steric repulsion to the preceding Cα atom than does the following Cα atom (cis isomer). By contrast, the cis and trans isomers of the X-Pro peptide bond are nearly isosteric (i.e., equally bad energetically); the Cα (cis isomer) and Cδ atoms (trans isomer) of proline are roughly equivalent sterically. Hence, the fraction of X-Pro peptide bonds in the cis isomer under unstrained conditions ranges from 10-40%; the fraction depends slightly on the preceding amino acid X, with aromatic residues favoring the cis isomer slightly.
Cis-trans proline isomerization is a very slow process that can impede the progress of protein folding by trapping one or more prolines crucial for folding in the nonnative isomer, especially when the native isomer is the rarer cis. All organisms possess prolyl isomerase enzymes to catalyze this isomerization, and some bacteria have specialized prolyl isomerases associated with the ribosome. However, not all prolines are essential for folding, and protein folding may proceed at a normal rate despite having non-native isomers of many X-Pro peptide bonds.
3. Synthesis and usage
Proline is biosynthetically derived from the amino acid L-glutamate and its direct precursor is the real imino acid (S)-Δ1-pyrroline-5-carboxylate (P5C).
Proline and its derivatives are often used as asymmetric catalysts in organic reactions. The CBS reduction or proline catalysed aldol condensation are prominent examples.
Proline has a sweet flavor with a distinct aftertaste. Proline also causes slight irritation to the tongue like Sichuan Pepper.
For unknown reasons, L-proline is used as an ingredient in energy drinks such as “Sobe power fruit punch”.
4. References
• Balbach J, Schmid FX. (2000). Proline isomerization and its catalysis in protein folding. In Mechanisms of Protein Folding 2nd ed. Editor RH Pain. Oxford University Press.
Serine
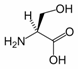
Serine is the organic compound with the formula HO2CCH(NH2)CH2OH. It is one of the 20 amino acids commonly found in animal proteins. Its three letter code is ser, its one letter code is S, and its codons are AGU and AGC.[1] Only the L-stereoisomer appears in mammalian protein. It is not essential to the human diet, since it is synthesized in the body from other metabolites, including glycine. Serine was first obtained from silk protein, a particularly rich source, in 1865. Its name is derived from the Latin for silk, sericum. Serine’s structure was established in 1902. The hydroxyl group attached makes it a polar amino acid.
1. Biosynthesis
The synthesis of serine starts with the oxidation of 3-phosphoglycerate forming 3-phosphohydroxypyruvate and NADH. Reductive amination of this ketone followed by hydrolysis affords serine. Serine hydroxymethyltransferase catalyzes the reversible, simultaneous conversions of L-serine to glycine (retro-aldol cleavage) and 5,6,7,8-tetrahydrofolate to 5,10-methylenetetrahydrofolate (hydrolysis).[2]
2. Function
2.1. Metabolic
Serine is important in metabolism in that it participates in the biosynthesis of purines and pyrimidines. It is also the precursor to several amino acids, including glycine, cysteine, tryptophan (in bacteria). It is also the precursor to numerous of other metabolites, including sphingolipids. Serine is also a precursor to folate which is the principal donor of one carbon fragments in biosynthesis.
2.2. Structural
Serine plays an important role in the catalytic function of many enzymes. It has been shown to occur in the active sites of chymotrypsin, trypsin, and many other enzymes. The so-called nerve gases and many substances used in insecticides have been shown to act by combining with a residue of serine in the active site of acetylcholine esterase, inhibiting the enzyme completely. Without the esterase activity that usually destroys acetylcholine as soon as it performs its function, dangerously high levels of this neurotransmitter build up, quickly resulting in convulsions and death.
As a constituent (residue) of proteins, its side chain can undergo O-linked glycosylation. This might be important in explaining some of the devastating consequences of diabetes. It is one of three amino acid residues that are commonly phosphorylated by kinases during cell signaling in eukaryotes. Phosphorylated serine residues are often referred to as phosphoserine. Serine proteases are a common type of protease.
2.3. Signaling
D-serine, synthesized by serine racemase from L-serine, serves as a neuronal signaling molecule by activating NMDA receptors in the brain.
3. Chemical Synthesis
Serine is prepared from methyl acrylate.[3]
4. References
1. IUPAC-IUBMB Joint Commission on Biochemical Nomenclature. Nomenclature and Symbolism for Amino Acids and Peptides. Recommendations on Organic & Biochemical Nomenclature, Symbols & Terminology etc.
2. Nelson, D. L.; Cox, M. M. “Lehninger, Principles of Biochemistry” 3rd Ed. Worth Publishing: New York, 2000. ISBN 1-57259-153-6.
3. Carter, H. E.; West, H. D. “dl-Serine” Organic Syntheses, Collected Volume 3, p.774 (1955).
Threonine
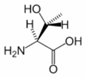
Threonine is an α-amino acid with the chemical formula HO2CCH(NH2)CH(OH)CH3. Its three letter code is thr, its one letter code is T, and its codons are ACU and ACA. This essential amino acid is classified as polar. Together with serine and tyrosine, threonine is one of three proteinogenic amino acids bearing an alcohol group.
The threonine residue is susceptible to numerous posttranslational modifications. The hydroxy side chain can undergo O-linked glycosylation. Additionally, threonine residues undergo phosphorylation through the action of a threonine kinase. In its phosphorylated form, it can be referred to as phosphothreonine.
1. Allo-threonine
With two chiral centers, threonine can exist in four possible stereoisomers, or two possible diastereomers of L-threonine. However, the name L-threonine is used for one single enantiomer, (2S,3R)-2-amino-3-hydroxybutanoic acid. The second diastereomer (2S,3S), which is rarely present in nature, is called L-allo-threonine.
2. Biosynthesis
As an essential amino acid, threonine is not synthesized in humans, hence we must ingest threonine or, more commonly, threonine-containing proteins. In plants and microorganisms, threonine is synthesized from aspartic acid via α-aspartyl-semialdehyde and homoserine. Homoserine undergoes O-phosphorylation; this phosphate ester undergoes hydrolysis concomitant with relocation of the OH group.[1] Enzymes involved in a typical biosynthesis of threonine include:
• aspartokinase
• α-aspartate semialdehyde dehydrogenase
• homoserine dehydrogenase
• homoserine kinase
• threonine synthase
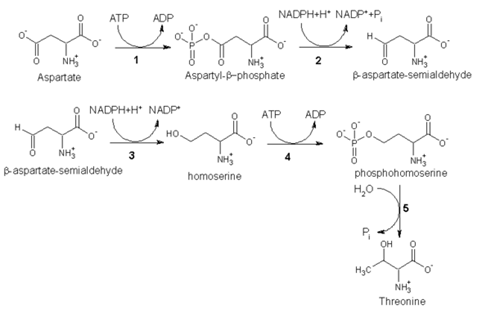
Threonine biosynthesis
3. Metabolism
Threonine is metabolized in two ways:
• It is converted to pyruvate
• It is converted to alpha-ketobutyrate, and thereby enter the pathway leading to succinyl CoA.
4. Synthesis
Racemic threonine can be prepared from crotonic acid by alpha-functionalization using mercury(II) acetate.[2]
5. Sources
Foods high in threonine include cottage cheese, poultry, fish, meat, lentils, and sesame seeds.
6. References
1. Nelson, D. L.; Cox, M. M. “Lehninger, Principles of Biochemistry” 3rd Ed. Worth Publishing: New York, 2000. ISBN 1-57259-153-6.
2. Carter, H. E.; West, H. D. (1955). Org. Synth.; Coll. Vol. 3: 813.
Tryptophan
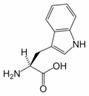
Tryptophan is an amino acid essential in human nutrition. It is one of the 20 amino acids encoded by the genetic code (as codon UGG). Only the L-stereoisomer appears in mammalian protein, however the D-stereoisomer is occasionally found in natural materials (for example, the marine venom peptide contryphan[1]). A distinquishing structural characteristic of tryptophan is that it contains an indole functional group.
Contents
1. Function
2. Dietary sources
3. Medicinal uses
4. Fluorescence
5. References
1. Function
For many organisms including humans, tryptophan is an essential amino acid. This means that it cannot be synthesized by the organism and therefore must be part of its diet. The principle function of amino acids including tryptophan are as building blocks in protein biosynthesis. In addition, tryptophan functions as a biochemical precursor for the following:
• Serotonin (a neurotransmitter), via tryptophan hydroxylase.
• Niacin (with kynurenine as an intermediate)
In organisms which synthesize tryptophan, high levels of this amino acid activate a repressor protein which in turn binds to the trp operon. Binding of this repressor to its operon prevents transcription of downstream DNA which codes for enzymes involved in the biosynthesis of tryptophan. Hence high levels of tryptophan prevent additional tryptophan synthesis through a negative feedback loop. Conversely if the cell’s tryptophan level drops, transcription of the operon’s genes resumes. This is one example of how gene expression responds rapidly to changes in the cell’s internal and external environment.
2. Dietary sources
Tryptophan, found as a component of dietary protein, is particularly plentiful in chocolate, oats, bananas, dried dates, milk, yogurt, cottage cheese, red meat, eggs, fish, poultry, sesame, chickpeas, sunflower seeds, pumpkin seeds, spirulina and peanuts.
3. Medicinal uses
5-Hydroxytryptophan (5-HTP), a metabolite of tryptophan, has been suggested as a treatment for epilepsy[1] and depression though clinical trials are inconclusive and lacking.[2]
5-HTP readily crosses the blood brain barrier and in addition is rapidly decarboxylated to serotonin (5-hydroxytryptamine or 5-HT)[3] and therefore may useful for the treatment of depression. However serotonin has a relatively short half life since it is rapidly metabolized by monoamine oxidase therefore is likely to have limited efficacy. It is marketed in Europe for depression and other indications under brand names like Cincofarm and Tript-OH.
In the United States, 5-HTP does not require a prescription as it is covered under the Dietary Supplement Act. However, since the quality of dietary supplements is not regulated by the FDA, the quality of dietary and nutritional supplements tends to vary and there is no guarantee that the label accurately depicts what the bottle contains.
In recent years, compounding pharmacies and some mail-order supplement retailers have begun selling tryptophan to the general public. Tryptophan has also remained on the market as a prescription drug (Tryptan) which some psychiatrists continue to prescribe, particularly as an augmenting agent for people who are unresponsive to antidepressant drugs. Also, most health-food stores sell 5-HTP to get around the resulting artificially high cost of the amino acid itself. But several high quality sources of L-Tryptophan do exist, and are sold in many of the largest health food stores nationwide. Indeed, tryptophan has continued to be used in clinical and experimental studies employing human patients and subjects. Several of these studies suggest tryptophan can effectively treat the fall/winter depression variant of seasonal affective disorder.[4]
4. Fluorescence
The fluorescence of a folded protein is a mixture of the fluorescence from individual aromatic residues. Most of the intrinsic fluorescence emissions of a folded protein are due to excitation of tryptophan residues, with some emissions due to tyrosine and phenylalanine. Typically, tryptophan has a wavelength of maximum absorption of 280 nm and a wavelength of maximum fluorescence emission of 350 nm. However these fluorescence parameters are strongly dependent on the environment that the tryptophan residue is in, for example the degree of solvent exposure.[5] Hence protein fluorescence may be used as a diagnostic of the conformational state a protein.[6]
Furthermore, tryptophan fluorescence is strongly influenced by the proximity of other residues (i.e., nearby protonated acidic groups such as Asp or Glu can cause quenching of Trp fluorescence). In addition, tryptophan is a relatively rare amino acid therefore many proteins contain only one or a few tryptophan residues. Therefore, tryptophan fluorescence is a very sensitive measurement of the conformational state of individual tryptophan residues.
5. References
1. Kostowski W, Bidzinski A, Hauptmann M, Malinowski JE, Jerlicz M, Dymecki J (1978). “Brain serotonin and epileptic seizures in mice: a pharmacological and biochemical study”. Pol J Pharmacol Pharm 30 (1): 41-7.
2. Turner EH, Loftis JM, Blackwell AD (2006). “Serotonin a la carte: supplementation with the serotonin precursor 5-hydroxytryptophan”. Pharmacol Ther 109 (3): 325-38.
3. Hardebo JE, Owman C (1980). “Barrier mechanisms for neurotransmitter monoamines and their precursors at the blood-brain interface”. Ann NeurolAnn Neurol 8 (1): 1-31.
4. Jepson TL, Ernst ME, Kelly MW (1999). “Current perspectives on the management of seasonal affective disorder”. J Am Pharm Assoc (Wash) 39 (6): 822-9.
5. Intrinsic Fluorescence of Proteins and Peptides
6. Vivian JT, Callis PR (2006). “Mechanisms of tryptophan fluorescence shifts in proteins”. Biophys J 80 (5): 2093-109.
Tyrosine
Tyrosine, 4-hydroxyphenylalanine, or 2-amino-3(4-hydroxyphenyl)-propanoic acid, is one of the 20 amino acids that are used by cells to synthesize proteins. It has a phenol side chain with a hydroxyl group. Upon the location of the hydroxyl group, there are three structural isomers of Tyr, namely para-Tyr (p-Tyr), meta-Tyr (m-Tyr) and ortho-Tyr (o-Tyr). Enzymatically, only the first isomer (p-Tyr) is produced from L-Phe by the Phe-hydroxylase enzyme. The other two isoforms, m-Tyr and o-Tyr can be produced as a consequence of free radical attack on Phe in states with increased oxidative stress.
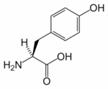
Tyrosine is converted to levodopa by tyrosine hydroxylase, an enzyme.
Some of the tyrosine residues can be tagged with a phosphate group (phosphorylated) by protein kinases. (In its phosphorylated state, it is referred to as phosphotyrosine.). Tyrosine phosphorylation is considered as one of the key steps in signal transduction and regulation of enzymatic activity. Phosphotyrosine can be detected through specific antibodies. Tyrosine residues may also be modified by the addition of a sulfate group, a process known as tyrosine sulfation. Tyrosine sulfation is catalyzed by tyrosylprotein sulfotransferase (TPST). Like the phosphotyrosine antibodies mentioned above, antibodies have recently been described that specifically detect sulfotyrosine.
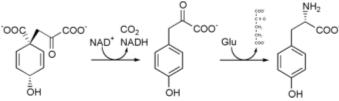
1. Biosynthesis
Tyrosine cannot be completely synthesized by animals, although it can be made by hydroxylation of phenylalanine if the latter is in abundant supply. It is produced by plants and most microorganisms from prephenate, an intermediate on the shikimate pathway.
Prephenate is oxidatively decarboxylated with retention of the hydroxyl group to give p-hydroxyphenylpyruvate. This is transaminated using glutamate as the nitrogen source to give tyrosine and α-ketoglutarate.
2. Tyrosine hydroxylase
Tyrosine hydroxylase (TH) is the rate-limiting enzyme involved in the synthesis of the catecholamines such as dopamine, norepinephrine and epinephrine.
3. Medical use
L-Tyrosine is sometimes recommended by practitioners as helpful for weight loss, clinical depression, Parkinson’s Disease, and phenylketonuria; however, one study found that it had no impact on endurance exercise performance. [1]
4. References
1. Parcell A.C., et al. Effects of L-tyrosine and carbohydrate ingestion on performance. Journal of Applied Physiology 2002 Nov; 93(5): 1590-97. Abstract
Valine
Valine is an α-amino acid with the chemical formula HO2CCH(NH2)CH(CH3)2. Its three letter code is Val, its one letter code is V, and its codons are GUU, GUC, GUA, and GUG. This essential amino acid is classified as nonpolar. Along with leucine and isoleucine, valine is a branched-chain amino acid. It is named after the plant valerian. In sickle-cell disease, valine substitutes for the hydrophilic amino acid glutamic acid in hemoglobin. Because valine is hydrophobic, the hemoglobin does not fold correctly.
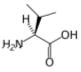
1. Biosynthesis
As an essential amino acid, valine is not synthesized in animals, hence it must be ingested, usually as a component of proteins. It is synthesized in plants via several steps starting from pyruvic acid. The initial part of the pathway also leads to leucine. The intermediate α-ketovalerate undergoes reductive amination with glutamate. Enzymes involved in this biosynthesis include:[1]
• acetolactate synthase
• acetohydroxy acid isomeroreductase
• dihydroxyacid dehydratase
• valine aminotransferase
2. References
1. Nelson, D. L.; Cox, M. M. “Lehninger, Principles of Biochemistry” 3rd Ed. Worth Publishing: New York, 2000. ISNB 1-57259-153-6.
