
Overview of Peptide Synthesis
This entry is from Wikipedia, the leading user-contributed encyclopedia.
In organic chemistry, peptide synthesis is the creation of peptides, which are organic compounds in which multiple amino acids bind via peptide bonds which are also known as amide bonds.
Contents
1. Chemistry
2. Liquid-phase synthesis
3. Solid-phase synthesis
3.1. Boc SPPS
3.2. Fmoc SPPS
4. Solid supports
4.1. Polystyrene resin
4.2. Polyamide resin
5. Protective groups
5.1. Fmoc protective group
5.2. Boc protective group
5.3. Benzyloxy-carbonyl (Z) group
5.4. Alloc protecting group
5.5. Lithographic protecting groups
6. Activating groups
6.1. Carbodiimides
6.2. Aromatic oximes
7. Synthesizing long peptides
8. An example of solid phase peptide synthesis
9. References
1. Chemistry
Peptides are synthesized by coupling the carboxyl group or C-terminus of one amino acid to the amino group or N-terminus of another.
2. Solution-phase synthesis
Solution-phase peptide synthesis is a classical approach to peptide synthesis. It has been replaced in most labs by solid-phase synthesis (see below). However, it retains usefulness in large-scale production of peptides for industrial purposes.
3. Solid-phase synthesis
Solid-phase peptide synthesis (SPPS), pioneered by Merrifield, resulted in a paradigm shift within the peptide synthesis community. It is now the accepted method for creating peptides and proteins in the lab in a synthetic manner. SPPS allows the synthesis of natural peptides which are difficult to express in bacteria, the incorporation of unnatural amino acids, peptide/protein backbone modification, and the synthesis of D-proteins, which consist of D-amino acids.

The solid phase peptide synthesis (SPPS) principle
Small beads are treated with linkers on which peptide chains can be built. The synthesis beads will retain strong bondage to the peptides until cleaved by a reagent such as trifluoroacetic acid. The beads create a synthesis environment in which the peptide chains being created will not pass through a filter material while the reagents used to create them will.
The overwhelmingly important consideration is to generate extremely high yield in each step. For example, if each step were to have 99% yield, a 26-amino acid peptide would be synthesized in 77% final yield, if each step were 95%, it would be synthesized in 25% yield. Thus each amino acid is added in major excess (2~10x) and coupling amino acids together is highly optimized by a series of well-characterized agents
There are two majorly used forms of SPPS – Fmoc and Boc. Unlike ribosome protein synthesis, solid-phase peptide synthesis proceeds in a C-terminal to N-terminal fashion. The N-termini of amino acid monomers is protected by these two groups and added onto a deprotected amino acid chain.
Automated synthesizers are available for both techniques, though many research groups continue to perform SPPS manually.
SPPS is limited by yields, and typically peptides and proteins in the range of 70~100 amino acids are pushing the limits of synthetic accessibility. Synthetic difficulty also is sequence dependent; typically amyloid peptides and proteins are difficult to make. Longer lengths can be accessed by using native chemical ligation to couple two peptides together with quantitative yields.
3.1. Boc SPPS
When R. B. Merrifield invented SPPS in 1963, it was according to the tBoc method. t-Boc (or Boc) stands for tert-Butyloxycarbonyl. To remove Boc from a growing peptide chain, acidic conditions are used (usually neat TFA). Removal of side-chain protecting groups and the peptide from the resin at the end of the synthesis is achieved by incubating in hydrofluoric acid (which can be dangerous); for this reason Boc chemistry is generally disfavored. However for complex syntheses Boc is favourable. When synthesizing nonnatural peptide analogs which are base-sensitive (such as depsi-peptides), Boc is necessary.
3.2. Fmoc SPPS
This method was introduced by R.C. Sheppard in 1971. Fmoc stands for 9-Fluorenylmethoxycarbonyl which describes the Fmoc protecting group. To remove an Fmoc from a growing peptide chain, basic conditions (usually 20% piperidine in DMF) are used. Removal of side-chain protecting groups and peptide from the resin is achieved by incubating in trifluoroacetic acid (TFA). Fmoc deprotection is usually slow because the anionic nitrogen produced at the end is not a particularly favorable product, although the whole process is thermodynamically driven by the evolution of carbon dioxide. The main advantage of Fmoc chemistry is that no hydrofluoric acid is needed. It is therefore used for most routine synthesis.
4. Solid supports
The physical properties of the solid support, and the applications to which it can be utilized, vary with the material from which the support is constructed, the amount of crosslinking, as well as the linker and handle being used.
4.1. Polystyrene resin
This is a versatile resin, which is quite useful in multi-well, automated peptide synthesis, due to its minimal swelling in DCM.
4.2. Polyamide resin
This too is a useful and versatile resin. It seems to swell much more than polystyrene, in which case it may not be suitable for some automated synthesizers, if the wells are too small.
5. Protective groups
Due to amino acid excesses used to ensure complete coupling during each synthesis step, polymerization of amino acids is common in reactions where each amino acid is not protected. In order to prevent this polymerization, protective groups are used. This adds additional deprotection phases to the synthesis reaction, creating a repeating design flow as follows:
• Protective group is removed from trailing amino acids in a deprotection reaction
• Deprotection reagents washed away to provide clean coupling environment
• Protected amino acids dissolved in a solvent such as dimethylformamide (DMF) are combined with coupling reagents are pumped through the synthesis column
• Coupling reagents washed away to provide clean deprotection environment
Currently, two protective groups (Fmoc, Boc) are commonly used in solid-phase peptide synthesis. Their lability is caused by the carbamate group which readily releases CO2 for an irreversible decoupling step.
5.1. Fmoc protective group
The Fmoc (9-fluorenylmethyl carbamate) is currently a widely used protective group that is generally removed from the N terminus of a peptide in the iterative synthesis of a peptide from amino acid units. The advantage of Fmoc is that it is cleaved under very mild basic conditions (e.g. piperidine), but stable under acidic conditions. This allows mild acid labile protecting groups that are stable under basic conditions, such as Boc and benzyl groups, to be used on the side-chains of amino acid residues of the target peptide. This orthogonal protecting group strategy is common in the art of organic synthesis.
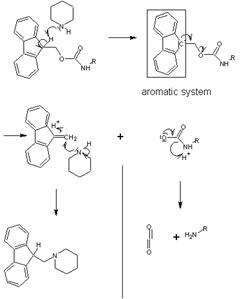
Fmoc cleavage
5.2. Boc protective group
Before the Fmoc group became popular, the Boc group was commonly used for protecting the terminal amine of the peptide, requiring the use of more acid stable groups for side chain protection in orthogonal strategies. It retains usefulness in reducing aggregation of peptides during synthesis. Boc groups can be added to amino acids with boc anhydride and a suitable base.
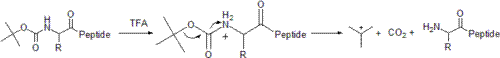
Boc cleavage
5.3. Benzyloxy-carbonyl (Z) group
Another carbamate based group is the benzyloxy-carbonyl (Z) group. It is removed in harsher conditions: HBr/acetic acid or catalytic hydrogenation. Today it is almost exclusively used for side chain protection.
5.4. Alloc protecting group
The allyloxycarbonyl (alloc) protecting group is often used to protect a carboxylic acid, hydroxyl, or amino group when an orthagonal deprotection scheme is required. It is sometimes used when conducting on-resin cyclic peptide formation, where the peptide is linked to the resin by a side-chain functional group. The alloc group can be removed using tetrakis(triphenylphosphine)palladium(0) along with a 37:2:1 mixture of chloroform, acetic acid, and N-methylmorpholine (NMM) for 2 hours. The resin must then be carefully washed 0.5 % DIPEA in DMF, 3×10 ml of 0.5 % sodium diethylthiocarbamate in DMF, and then 5×10 ml of 1:1 DCM:DMF.
5.5. Lithographic protecting groups
For special applications like protein microarrays lithographic protecting groups are used. Those groups can be removed through exposure to light.
6. Activating groups
For coupling the peptides the carboxyl group is usually activated. This is important for speeding up the reaction. There are two main types of activating groups: carbodiimides and aromatic oximes.
6.1. Carbodiimides
These activating agents were first developed. Most common are dicyclohexylcarbodiimide (DCC) and diisopropylcarbodiimide (DIC). Reaction with a carboxylic acid yields a highly reactive O-acyl-urea. During artificial protein synthesis (such as Fmoc solid-state synthesizers), the C-terminus is often used as the attachement site on which the amino acid monomers are added. To enhance the electrophilicity of carboxylate group, the negatively charged oxygen must first be “activated” into a better leaving group. DCC is used for this purpose. The negatively charged oxygen will act as a nucleophile, attacking the central carbon in DCC. DCC is temporarily attached to the former carboxylate group (which is now an ester group), making nucleophilic attack by an amino group (on the attaching amino acid) to the former C-terminus(carbonil group) more efficient. The problem with carbodiimides is that they are too reactive and that they can therefore cause racemization of the amino acid.
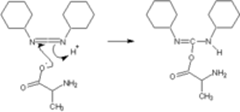
Alanine attaching to DCC
6.2. Aromatic oximes
To solve the problem of racemization, aromatic oximes were introduced. The most important ones are 1-hydroxy-benzotriazole (HOBt) and 1-hydroxy-7-aza-benzotriazole (HOAt).
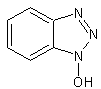
HOAt
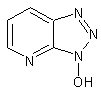
HOBt
Others have been developed. These substances can react with the O-acylurea to form an active ester which is less reactive and less in danger of racemization. HOAt is especially favourable because of a neighbouring group effect. [1]
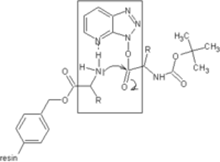
Neighbouring group effect of HOAt
Newer developments omit the carbodiimides totally. The active ester is introduced as an uronium or phosphonium salt of a non-nucleophilic anion (tetrafluoroborate or hexafluorophosphate): HBTU, HATU, PyBOP.
7. Synthesizing long peptides
Stepwise elongation, in which the amino acids are connected step-by-step in turn, is ideal for small peptides containing between 2 and 100 amino acid residues. Another method is fragment condensation, in which peptide fragments are coupled. Although the former can elongate the peptide chain without racemization, the yield drops if only it is used in the creation of long or highly polar peptides. Fragment condensation is better than stepwise elongation for synthesizing sophisticated long peptides, but its use must be restricted in order to protect against racemization. Fragment condensation is also undesirable since the coupled fragment must be in gross excess, which may be a limitation depending on the length of the fragment.
A new developement for producing longer peptide chains is chemical ligation: Unprotected peptide chains react chemoselectively in aqueous solution. A first kinetically controlled product rearranges to form the amide bond. The most common form native chemical ligation uses a peptide thioester that reacts with a terminal cystein residue.
8. An example of solid phase peptide synthesis
The following is an outline of the synthetic steps for peptide synthesis on polyamide or polystyrene resin, using the base labile 9-fluorenylmethyloxycarbonyl (Fmoc) protecting group. Using the techniques outlined below, one will obtain a peptide which is capped on the N-terminus with and acetyl group, and on the C-terminus with a primary amide (CONH2).
Setting up glassware for manual peptide synthesis
Manual peptide synthesis can be accomplished in a fritted-filter reaction vessel with a three-way valve fitted onto a 1 L side arm vacuum flask by way of a 1-hole stopper. One valve is used to bubble nitrogen, which is first passed through a small column of Drierite, and then into the reaction mixture to agitate the solution and mix reagents. The other valve is used to evacuate excess reaction solutions and wash solvent using a vacuum flask. All glass pieces to be used in Solid-phase synthesis should be treated with a silanizing agent (such as 1-5% dimethyldichlorosilane in DCM) prior to use, to avoid accumulation of static charge, which makes the resin very difficult to handle.
Preparation of polyamide-Rink resin
Polyamide (PL-DMA) resin (1g) is treated with ethylene diamine (40 ml) in a 50 ml Falcon tube overnight on a rocker, then filtered, washed with 5×10 ml of 1:1 dimethylformamide (DMF):dichloromethane (DCM) solution, 5×10 ml of 1:1 DCM, and loaded with Fmoc-Rink using Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) (3 eq), 1-Hydroxybenzotriazole Hydrate (HOBt) (3 eq), and Diisopropylethylamine (DIPEA) (6 eq) in 1:1 DCM:DMF. It can then be dried under vacuum and stored at -15oC until needed.
Handling the resin before, and during synthesis
The resin is first swelled for 15 minutes in 10 ml of 1:1 DCM:DMF and drained. The resin is also washed with 5×10 ml of 1:1 DCM and DMF after each completed amino acid coupling.
Fmoc-deprotection
Fmoc deprotection after each amino acid coupling is accomplished using 2×10 ml of 20% piperidine in DMF, with N2 agitation for 10 minutes each treatment. The resin is then washed with 5×10 ml DMF, followed by 5×10 ml of 1:1 DCM:DMF.
Adding amino acids
To begin peptide synthesis on Fmoc-polyamide-Rink resin the Fmoc group is by removed by treating the resin with piperidine. The first amino acid is then coupled to the Fmoc-deprotected N-terminal amine of the resin, or previously coupled amino acid, using PyBOP (3 eq), HOBt (3 eq), and DIPEA (6 eq) in 1:1 DCM:DMF until the resin is negative to ninhydrin.
Monitoring the progress of amino acid couplings
The progress of amino acid couplings can be followed using ninhydrin, or p-chloranil. The ninhydrin solution turns dark blue (positive result) in the presence of a free primary amine but is otherwise colorless (negative result). The p-chloranil solution will turn the resin beads dark black or blue in the presence of a primary amine if acetaldehyde is used as the solvent or in the presence of a secondary amine, if acetone is used instead; the beads remain colorless or pale yellow otherwise. (The tests are outlined below)
Testing by Ninhydrin (1)
Add 2 drops of 40% phenol in ethanol, 2 drops of 0.014 M KCN in pyridine, and 4 drops of 5% ninhydrin in ethanol to a microcentrifuge tube along with a spatula tip size sample of resin, then vortex the mixture and heat for 5 minutes at 100 °C.
Testing by Chloranil (2)
Add 5 drops of acetone or acetaldehyde, 5 drops of a saturated solution of p-chloranil in toluene, plus a small spatula-tip-size sample of resin to a microcentrifuge tube, then vortex the mixture and allow to stand at room temperature for 5 minutes. Acetone is used for the detection of secondary amines, where acetaldehyde is used for primary amines.
Continuing peptide extension
Once the coupling of the amino acid is complete, the resin is washed, the Fmoc group deprotected with piperidine, and the resin washed again to prepare it for the next coupling. This process is repeated until all necessary amino acids have been added.
Acetylating the N-terminus
After the peptide sequence is completed, the N-terminal amine can be acetylated with 2 ml of 1:1 acetic anhydride and triethylamine in 10 ml of 1:1 DCM:DMF for 1 hour or until negative to ninhydrin, and the resin then washed with 5×10 ml of 1:1 DCM:DMF, before the peptide is cleaved from the resin. The N-terminal can also be left as the free amine if required.
Cleaving the peptide from the resin
The resin is treated with 3×10 ml of 96:2:2 trifluoroacetic acid (TFA), triisopropylsilane (TIPS), and water (H2O) for 10 min each treatment, the resin is then filtered away, and the combined filtrates allowed to stand for 1 hour to ensure removal of the acid labile protecting groups.
Workup of peptides after cleavage from the resin
The TFA is evaporated to dryness (or a heavy oil or glass if it does not solidify) on the rotary evaporator, followed by the addition of 5 ml of diethyl ether to the flask to precipitate the peptide, and remove the bulk of the by-products. The suspended mixture of peptide and ether is added to a 50 ml Falcon tube and spun at 3000 rpm for 10–20 min (IEC centrifuge) until the ether can be decanted off without losing any peptide. More ether is added, and the peptide resuspeded by vortexing. The mixture is centrifuged again, the ether decanted, and the washing process repeated twice more. Finally the product can be air dried overnight in the Falcon tube in a descicator.
Typically preparative HPLC is used to purify the final product. Mass spectrometry data is obtained to ensure the target peptide was obtained.
9. References
• Atherton, E., Sheppard, R.C. Solid phase peptide synthesis: a practical approach. IRL Press, Oxford, England, 1989.
• Stewart J.M., Young, J.D. Solid phase peptide synthesis, 2nd edition, Pierce Chemical Company, Rockford, 1984, pp 91.
1. Carpino, L. A. 1992 1-Hydroxy-7-azabenzotriazole. An efficient Peptide Coupling Additive. J. Am. Chem. Soc. 115, 4397-4398.
