
Reagents & Reactions
This entry is from Wikipedia, the leading user-contributed encyclopedia.
• Biotin
• Carbodiimide
• t-Boc
• Ninhydrin
• Piperidine
• DMAP
• DIPEA
• Trifluoroacetic acid
• Edman degradation
• Chemical ligation
• Native chemical ligation
• PEG
• PEGylation
• Freeze drying
Biotin
Biotin, also known as vitamin H or B7, has the chemical formula C10H16N2O3S (Biotin; Coenzyme R, Biopeiderm), is a water-soluble B-complex vitamin which is composed of an ureido (tetrahydroimidizalone) ring fused with a tetrahydrothiophene ring. A valeric acid substituent is attached to one of the carbon atoms of the tetrahydrothiophene ring. Biotin is important in the catalysis of essential metabolic reactions to synthesize fatty acids, in gluconeogenesis, and to metabolize leucine.
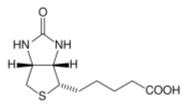
1. General overview
Biotin is used in cell growth, the production of fatty acids, metabolism of fats, and amino acids. It plays a role in the Citric acid cycle, which is the process in which biochemical energy is generated during aerobic respiration. Biotin not only assists in various metabolic chemical conversions, but also helps to transfer carbon dioxide. Biotin is also helpful in maintaining a steady blood sugar level. Biotin is often recommended for strengthening hair and nails. Consequently, it is found in many cosmetic and health products for the hair and skin.
Deficiency is extremely rare, as intestinal bacteria generally produce in excess of the body’s daily requirement. For that reason, statutory agencies in many countries (e.g., the Australian Department of Health and Aging) do not prescribe a recommended daily intake.
2. Biochemistry
Biotin is a cofactor responsible for carbon dioxide transfer in several carboxylase enzymes:
• Acetyl-CoA carboxylase alpha
• Acetyl-CoA carboxylase beta
• Methylcrotonyl-CoA carboxylase
• Propionyl-CoA carboxylase
• Pyruvate carboxylase
The attachment of biotin to various chemical sites, called biotinylation, can be used as an important laboratory technique to study various processes including protein localization, protein interactions, DNA transcription and replication. Biotin itself is known to biotinylate histones, but is not found naturally on DNA. Holocarboxylase synthetase is involved in the binding of biotin.
Biotin binds very tightly to the tetrameric protein avidin (also streptavidin and neutravidin), with a dissociation constant Kd in the order of 10-15 mol/L (Bonjour, 1977; Green 1975; and Roth, 1985). This is often used in different biotechnological applications. Until 2005, very harsh conditions were required to break the biotin-streptavidin bond.[1]
3. Laboratory uses
In the biology laboratory, biotin is sometimes chemically linked, or tagged, to a molecule or protein for biochemical assays. This process is called biotinylation. Since avidins bind preferentially to biotin, biotin-tagged molecules can be extracted from a sample by mixing them with beads with covalently-attached avidin, and washing away anything unbound to the beads.
For example, biotin can be attached to a molecule of interest (e.g. a protein), and this modified molecule will be mixed with a complex mixture of proteins. Avidin or streptavidin beads are added to the mixture, and the biotinylated molecule will bind to the beads. Any other proteins binding to the biotinylated molecule will also stay with the beads. All other unbound proteins can be washed away, and the scientist can use a variety of methods to determine which proteins have bound to the biotinylated molecule.
Biotinylated antibodies are used to capture avidin or streptavidin in both the ELISPOT and ELISA techniques.
4. References
1. Holmberg A, Blomstergren A, Nord O et al. (2005). “The biotin-streptavidin interaction can be reversibly broken using water at elevated temperatures”. Electrophoresis 26 (3): 501-10.
Carbodiimide
A carbodiimide is a functional group consisting of the formula N=C=N. Carbodiimides hydrolyze to form ureas, which makes them rarely found in nature.
Contents
1. Carbodiimide formation
2. Uses of carbodiimides
2.1. Amide formation mechanism
3. DCC
3.1. Structure
3.2. Synthesis of DCC
3.3. Reactivity of DCC
3.4. Mechanism of DCC-promoted peptide coupling
3.5. External links
3.6. References
4. DIC
4.1. References
4.2. External links
5. EDC
5.1. References
5.2. External links
1. Carbodiimide formation
Carbodiimides are formed by dehydration of ureas or from thioureas.
2. Uses of carbodiimides
In synthetic organic chemistry, compounds containing the carbodiimide functionality are dehydration agents and are often used to activate carboxylic acids towards amide or ester formation. Additives, such as N-hydroxybenzotriazole or N-hydroxysuccinimide, are often added to increase yields and decrease side reactions.
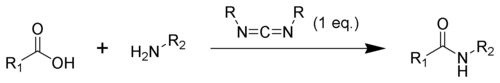
Carbodiimides can also react with amines to form guanidines.
2.1. Amide formation mechanism
The formation of an amide using a carbodiimide is straightforward, but with several side reactions complicating the subject. The acid 1 will react with the carbodiimide to produce the key intermediate: the O-acylisourea 2, which can be viewed as a carboxylic ester with an activated leaving group. The O-acylisourea will react with amines to give the desired amide 3 and urea 4.
The side reaction of the O-acylisourea 2 produce both desired and undesired products. The O-acylisourea 2 can react with an addition carboxylic acid 1 to give a carboxylic anhydride 5, which can react further to give the desired amide 3. The main undesired reaction pathway involves the rearrangement of the O-acylisourea 2 to the stable N-acylurea 6. The use of solvents with low-dielectric constants such as dichloromethane or chloroform can minimize this side reaction.
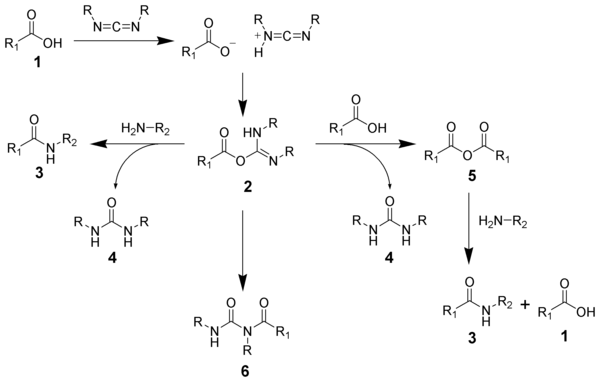
3. DCC
DCC (acronym for N,N’-dicyclohexylcarbodiimide) is an organic compound with chemical formula C13H22N2 whose primary use is to couple amino acids during artificial protein synthesis. Under standard conditions, DCC exists in the form of white crystals with a heavy, sweet odor. The low melting point of this material allows it to be melted for easy handling. DCC is highly soluble in dichloromethane, tetrahydrofuran, acetonitrile and dimethylformamide, but insoluble in water.
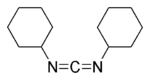
It should be handled with caution because it is a potent allergen and a sensitizer, often causing allergic reactions, particularly skin rashes.
3.1. Structure
The structure of DCC is not a planar structure as shown in the simplified picture above. Two resonance structures are available to DCC and elucidate the structure of this molecule:
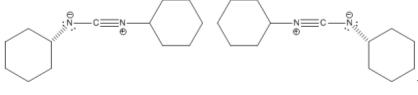
DCC resonance structures
These structures show that the central N=C=N moiety does remain linear; however, the cyclohexyl groups are not confined to a particular geometry. Also, a lack of pi bonding between the two nitrogens and the cyclohexyl groups allows them to rotate on the N-C bond axis.
3.2. Synthesis of DCC
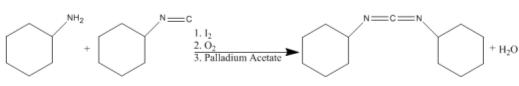
Of the several syntheses of DCC, Pri-Bara et. al. use palladium acetate, iodine, and oxygen to couple cyclohexyl amine and cyclohexyl isocyanide. Yields of up to 67% have been achieved using this reaction scheme. The reaction is as follows:
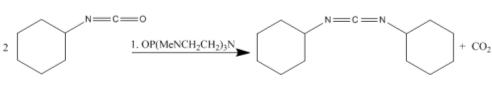
Tang et. al. condense two isocyanides using the catalyst OP(MeNCH2CH2)3N in yields of 92%. The reaction is as follows:
Lastly, a method for the synthesis of DCC involving a phase transfer catalyst has been published by Jaszay et. al. The disubstituted, arenesulfonyl chloride, and potassium carbonate react in the presence of benzyl trimethylammonium chloride.

The N=C=N moiety gives characteristic IR spectroscopic signature at 2117 cm-1 (Tang et. al.). 15N NMR shows a characteristic shift of 275.0 ppm upfield of nitric acid. 13C NMR of this compound contains a peak at about 139 ppm downfield from TMS depending on solvent choice (DMSO or Chloroform).
3.3 Reactivity of DCC
DCC is a strong dehydrating agent for the preparation of amides, ketones, nitriles, and can invert secondary alcohols. During reaction, it hydrates to form dicyclohexylurea (DCU), an insoluble compound.
Moffatt Oxidation
A solution of DCC and dimethylsulfoxide (DMSO) is used in a reaction termed the Pfitzner-Moffatt oxidation. This procedure is used for the gentle oxidation of alcohols to aldehydes and ketones. Unlike metal-mediated oxidations, the reaction conditions are sufficiently mild to halt over-oxidation of aldehydes to carboxylic acids. Generally, 1 equivalent of the alcohol to be oxidized is mixed with 3 equiv DCC and a proton source (0.5 equiv) in DMSO and left to react overnight at room temperature. The reaction is quenched by the addition of acid.
Primary alcohols:
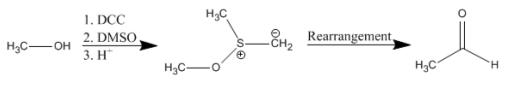
Secondary alcohols:
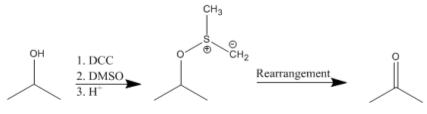
Dehydroxylation
Alcohols can also be dehydroxylated using DCC. This reaction proceeds by first forming an O-acylurea intermediate which is then hydrogenolyzed to produce the corresponding alkane.
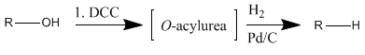
Inversion of secondary alcohols
Secondary alcohols can be stereochemically inverted by formation of a formyl ester followed by saponification. The secondary alcohol is mixed directly with DCC, formaldehyde, and a strong base such as sodium methoxide.
Esterification
A range of alcohols, including even some tertiary alcohols, can be esterified using a carboxylic acid in the presence of DCC and a catalytic amount of DMAP.
3.4. Mechanism of DCC-promoted peptide coupling
During artificial protein synthesis (such as Fmoc solid-state synthesizers), the C-terminus is often used as the attachement site on which the amino acid monomers are added. To enhance the electrophilicity of carboxylate group, the negatively charged oxygen must first be “activated” into a better leaving group. DCC is used for this purpose. The negatively charged oxygen will act as a nucleophile, attacking the central carbon in DCC. DCC is temporarily attached to the former carboxylate group (which is now an ester group), making nucleophilic attack by an amino group (on the attaching amino acid) on the former C-terminus more efficient.
3.5. External links
3.6. References
• Ilan Pri-Bara and Jeffrey Schwartz (1997). “N,N-Dialkylcarbodiimide synthesis by palladium-catalysed coupling of amines with isonitriles”. Chem Commun 4.
• Jiansheng Tang, Thyagarajan Mohan, John G. Verkade (1994). “Selective and Efficient Syntheses of Perhydro-1 ,3,5-triazine-2,4,6-trioneasn d Carbodiimides from Isocyanates Using ZP(MeNCH2CH2)sN Catalysts”. J. Org. Chem. 59: 4931-4938.
• Issa Yavari, John D. Roberts (1978). “Nitrogen-15 Nuclear Magnetic Resonance Spectroscopy. Carbodiimides”. J. Org. Chem. 43: 4689-4690.
• Zsuzsa Jaszay, Imre Petnehazy, Laszlo Toke, Bela Szajani (1987). “Preparation of Carbodiimides Using Phase-Transfer Catalysis”. Synthesis 5: 520-523.
4. DIC
DIC (acronym for N,N’-diisopropylcarbodiimide) was developed as an alternative to DCC. DIC is identical to DCC in nearly every way except:
1. As a liquid, DIC is easier to handle than DCC (which is a waxy solid).
2. The product, N,N’-diisopropylurea, is soluble in organic solvents and is easily removed by extraction. Hence, DIC is more often used in solid-phase synthesis.
3. Most importantly, DIC will not induce an allergic reaction.
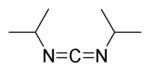
Structure of DIC
4.1. References
• Tetrahedron Lett. 1994, 35, 5981.
• Int. J. Pept. Protein Res. 1994, 43, 184.
4.2. External links
5. EDC
EDC (acronym for 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride) is a water soluble carbodiimide which is typically employed in the 4.0-6.0 pH range. It is generally used as a carboxyl activating agent for the coupling of primary amines to yield amide bonds. Additionally, EDC can also be used to activate phosphate groups. Commons uses for this carbodiimide include peptide synthesis, protein crosslinking to nucleic acids and preparation of immunoconjugates.
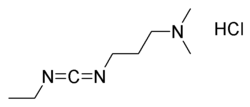
Structure of EDC.HCl
5.1. References
• Nakajima, N; Ikada, Y; Bioconjug Chem. 1995, 6(1), 123-130.
5.2. External links
• PubChem summary of EDC
• Carbodiimide Crosslinker Chemistry
Di-tert-butyl dicarbonate
Di-tert-butyl dicarbonate is a reagent widely used in organic synthesis.[1] This carbonate ester reacts with amines to give N-tert-butoxycarbonyl or so-called t-Boc derivatives. These derivatives do not behave as amines, which allow certain subsequent transformations to occur that would have otherwise affected the amine functional group. The t-Boc can later be removed from the amine using acids. Thus, t-Boc serves as a protective group, for instance in solid phase peptide synthesis. It is stable to most bases and nucleophiles, allowing for an orthogonal Fmoc protection.
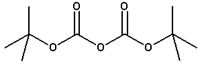
Structure of Di-tert-butyl dicarbonate
1. Protection and deprotection of amines
The Boc group can be added to the amine under aqueous conditions using di-tert-butyl dicarbonate in the presence of a base such as sodium bicarbonate. Protection of the amine can also be accomplished in acetonitrile solution using 4-dimethylaminopyridine (DMAP) as the base.
Removal of the t-Boc in amino acids can be accomplished with strong acids such as trifluoroacetic acid neat or in dichloromethane, or with HCl in methanol.[2] [3]
2. Other uses
The synthesis of 6-acetyl-1,2,3,4-tetrahydropyridine, an important bread aroma compound from 2-piperidone was accomplished using t-boc anhydride.[4] (See Maillard reaction). The first step in this reaction sequence is the formation of the carbamate from the reaction of the secondary amine with boc anhydride in acetonitrile with DMAP as a base.
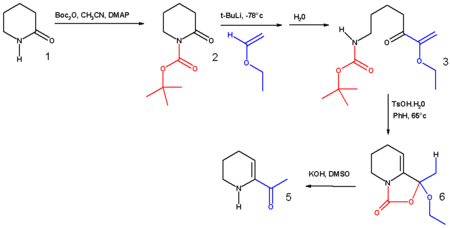
3. Preparation
Classically, this compound is prepared from tert-butanol, carbon dioxide, phosgene, using DABCO as a base:[5]
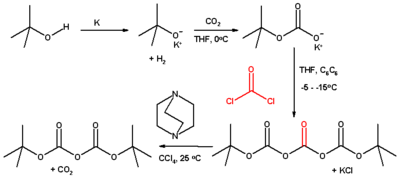
4. References
1. Wakselman, M. “Di-t-butyl Dicarbonate” in Encyclopedia of Reagents for Organic Synthesis (Ed: L. Paquette) 2004, J. Wiley & Sons, New York.
2. Robert M. Williams, Peter J. Sinclair, Duane E. DeMong, Daimo Chen, and Dongguan Zhai, Asymmetric Synthesis of N-tert-butoxycarbonyl α-Amino Acids, Organic Syntheses, Vol. 80, p.18 (2003)
3. E. A. Englund, H. N. Gopi, D. H. Appella, Org. Lett., 2004, 6, 213-215. DOI:10.1021/ol0361599
D. M. Shendage, R. Fröhlich, G. Haufe, Org. Lett., 2004, 6, 3675-3678.
4. Tyler J. Harrison and Gregory R. Dake, An Expeditious, High-Yielding Construction of the Food Aroma Compounds 6-Acetyl-1,2,3,4-tetrahydropyridine and 2-Acetyl-1-pyrroline, J. Org. Chem., 2005; 70(26) pp 10872 – 10874.
5. Barry M. Pope, Yutaka Yamamoto, and D. Stanley Tarbell, Dicarbonic acid, bis(1,1-dimethylethyl) ester, Organic Syntheses, Coll. Vol. 6, p.418 (1988); Vol. 57, p.45 (1977).
Ninhydrin
Ninhydrin, (Triketohydrindane hydrate) is a chemical used to detect ammonia or primary and secondary amines. When reacting with these free amines, a deep blue or purple color is evolved. Ninhydrin is most commonly used to detect fingerprints, as amines left over from peptides and proteins (terminal amines or lysine residues) sloughed off in fingerprints react with ninhydrin.[1]
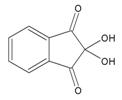
Structure of Ninhydrin
1. Uses
Ninhydrin can also be used to monitor deprotection in solid phase peptide synthesis (Kaiser Test).[2] When the growing peptide chain is deprotected, a ninhydrin test yields blue. If the next peptide residue is coupled then the test is colorless or yellow.
Ninhydrin is also used in amino acid analysis of proteins: Most of the amino acids are hydrolyzed and reacted with ninhydrin except Proline; Also, certain amino acid chains are degraded. Therefore, separate analysis is required for identifying such amino acids that either react differently or don’t react at all with ninhydrin. The rest of the amino acids are then quantified colorimetrically after separation by chromatography.
A solution suspected of containing the ammonium ion can be tested by ninhydrin by dotting it onto a solid support (such as silica gel); treatment with ninhydrin should result in a dramatic purple color if the solution contains this species.
2. Mechanism to generate the ninhydrin chromophore
Note that in order to generate the ninhydrin chromophore, the amine is condensed with a molecule of ninhydrin to give a Schiff base. Thus only ammonia and primary amines can proceed past this step. At this step, there must also be an alpha proton (H* in the diagram) for Schiff base transfer, so an amine adjacent to a tertiary carbon cannot be detected by the ninhydrin test. The reaction of ninhydrin with secondary amines gives an iminium salt, which is also coloured, and this is generally yellow-orange in color.
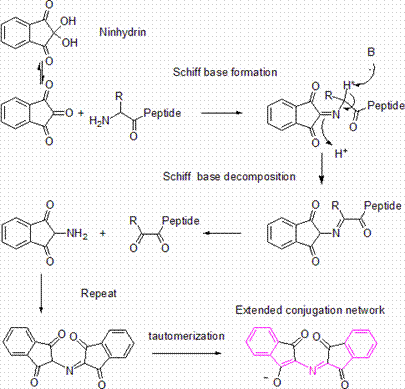
3. References
2. E. Kaiser, R. L. Colescott, C. D. Bossinger, P. I. Cook, Analytical Biochemistry 34, 595, 1970
4. External links
• Monitoring solid phase peptide synthesis
Piperidine
Piperidine is an organic compound with the molecular formula C5H11N. It is a heterocyclic amine with a six-membered ring containing five carbon atoms and one nitrogen atom. It is a clear liquid with a pepper-like odor.
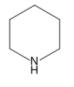
Structure of Piperidine
The piperidine structural motif is present in numerous natural alkaloids such as piperine and quinine, and is the main active chemical agent in black pepper and relatives (Piper sp.), hence the name. Piperidine is also a structural element of many pharmaceutical drugs such as raloxifene and minoxidil.
Piperidine is listed as a Table II precursor under the United Nations Convention Against Illicit Traffic in Narcotic Drugs and Psychotropic Substances [1] due to its use (peaking in the 1970s) in the clandestine manufacture of PCP. It is also a byproduct of burning phencyclidine (PCP, phenylcyclohexylpiperidine).
Piperidine is often used as a solvent for its mild basic properties, most notably in Fmoc-strategy solid phase peptide synthesis.
The major industrial application of piperidine is for the production of dipiperidinyl dithium tetrasulfide, which is used as a rubber vulcanization accelerator.
Piperidine is naturally found in fire ant venom, and is the cause of the burning sensation associated with the bite of these insects.
Piperide is also commonly used in chemical degradation reaction, one of the DNA sequencing method invented by Walter Gilbert in 1977, for cleavage of particular modified nucleotides.
DMAP
DMAP (4-Dimethylaminopyridine) is a nucleophilic catalyst which catalyzes a variety of reactions. For example esterifications with anhydrides, Baylis-Hillman reaction, silylation, tritylation, Steglich-Rearrangement, Staudinger synthesis of β-lactams and many more.[1][2]
Structure of DMAP
1. Esterification catalyst
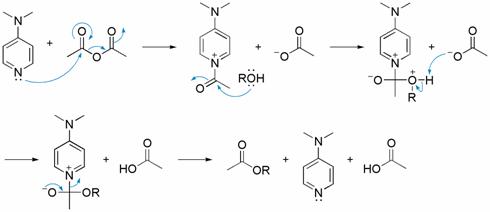
In the case of esterification with acetic anhydride the currently accepted mechanism involves three steps. First, DMAP and acetic anhydride form in an equilibrium reaction a labile ion pair between the acetate and the acetylpyridium ion. In the second step the alcohol attacks the acetyl group to form the ester. In this step the acetate counterion pulls away the proton from the alcohol while the alcohol forms a covalent bond with the acetyl group. The bond from the acetyl group to the catalyst gets cleaved to generate the catalyst and the ester. The acetic acid formed will then protonate the DMAP. In the last step of the catalytic cycle the auxiliary base (usually triethylamine) deprotonates the protonated DMAP, reforming the catalyst.[3]
2. References
1. Catalysis by 4-dialkylaminopyridines Donald J Berry, Charles V Digiovanna, Stephanie S Metrick and Ramiah Murugan Arkivoc O-401R 2001
2. 4-Dialkylaminopyridines as Highly Active Acylation Catalysts Höfle, G., Steglich, W., Vorbrüggen, H. Angew. Chem. Int. Ed. Engl. 1978, 17, 569-583.
3. The DMAP-Catalyzed Acetylation of Alcohols – A Mechanistic Study (DMAP=4-(dimethylamino)-pyridine) S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich, H. Zipse, Chem. Eur. J. 2005, 11, 4751 – 4757
DIPEA
N,N-Diisopropylethylamine, or Hünig’s base, DIPEA or DIEA, is an organic compound and an amine. It is used in organic chemistry as a base. Because the nitrogen atom is shielded by the two isopropyl groups and an ethyl group only a proton is small enough to fit. Just like 2,2,6,6-tetramethylpiperidine this compound is a base but not a nucleophile which makes it a special organic reagent.
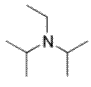
Structure of DIPEA
Hünig’s base forms a complex heterocyclic compound called Sscorpionine by a reaction with dichloro disulfide catalyzed by DABCO in a remarkable one-pot synthesis.[1]
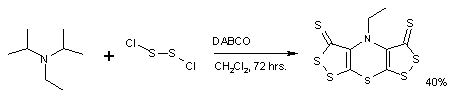
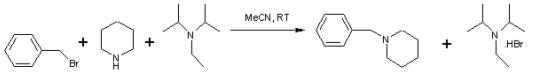
Hünig’s base is investigated for its use as a selective reagent in the alkylation of secondary amines to tertiary amines by alkyl halides. This organic reaction is often hampered by a quaternization reaction to the quaternary ammonium salt but this side-reaction is absent when Hünig’s base is present.[2]
References
1. From Hünig’s Base to Bis([1,2]dithiolo)-[1,4]thiazines in One Pot: The Fast Route to Highly Sulfurated Heterocycles W. Rees, Carlos F. Marcos,Cecilia Polo, Tomás Torroba,Oleg A. Rakitin Angewandte Chemie International Edition Volume 36, Issue 3 , Pages 281-283, 2003.
2. An efficient and operationally convenient general synthesis of tertiary amines by direct alkylation of secondary amines with alkyl halides in the presence of Huenig’s base Jason L. Moore, Stephen M. Taylor, and Vadim A. Soloshonok Arkivoc (EJ-1549C) pp 287-292, 2005.
Trifluoroacetic acid
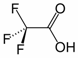
Trifluoroacetic acid (TFA) is the chemical compound with the formula CF3CO2H. It is a strong carboxylic acid due to the influence of the three very electronegative fluorine atoms. Relative to acetic acid, TFA is almost 100,000-fold more acidic. TFA is widely used in organic chemistry.
1. Uses
TFA is a reagent used frequently in organic synthesis because of a combination of convenient properties: volatility, solubility in organic solvents, and its strength.[1] It is also less oxidizing than sulfuric acid but more readily available in anhydrous form than hydrochloric acid. One complication to its use is that TFA forms an azeotrope with water with a boiling point of 105 °C.
It is also frequently used as a buffer in liquid chromatography for separation of organic compounds, particularly peptides and small proteins. It is a versatile solvent for NMR spectroscopy.
The derived acid anhydride, [CF3C(O)]2O, is a common reagent for introducing the trifluoracetyl group.
2. Synthesis
Electrofluorination of acetic acid with the Simons method is the best way to obtain trifluoroacetic acid. The anodic reaction of the electrolysis of a mixture of hydrogen fluoride and acetic acid below the voltage at which elemental fluorine (F2) develops is a mild reaction which leaves the carboxylic group intact.
3. References
1. Eidman, K. F.; Nichols, P. J. “Trifluoroacetic Acid” in Encyclopedia of Reagents for Organic Synthesis (Ed: L. Paquette) 2004, J. Wiley & Sons, New York.
Edman degradation
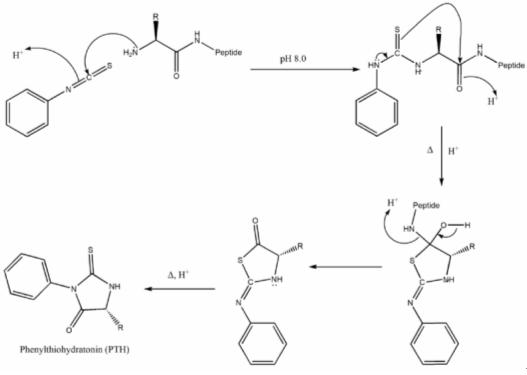
Edman degradation, developed by Pehr Edman, is a method of sequencing amino acids in a peptide. In this method, the amino-terminal residue is labeled and cleaved from the peptide without disrupting other peptide bonds between other amino acid residues. Phenylisothiocyanate is reacted with uncharged terminal amino group, under mildly alkaline conditions, to form a phenylthiocarbamoyl derivative. Then, under acidic conditions, this derivative of the terminal amino acid is cleaved as a thiazolinone derivative. The thiazolinone amino acid is then selectively extracted into an organic solvent and treated with acid to form the more stable phenylthiohydantoin (PTH) – amino acid derivative that can be identified by using chromatography or electrophoresis. This procedure can then be repeated again to identify the next amino acid. A major drawback to this technique is that the peptides being sequenced in this manner cannot have more than 50 to 60 residues. This is because the Edman degradation reaction is not 100% efficient, meaning that the cleavage step does not occur every time. However, this problem can be resolved by cleaving large peptides into smaller peptides before proceeding with the reaction. It is able to accurately sequence up to 30 amino acids with 98% efficiency per amino acid. An advantage of the Edman degradation is that it only uses 10 – 100 picomoles of peptide for the sequencing process.
1. Mechanism for Edman Degradation
2. Bergmann Degradation
An older related method for the stepwise degradation of polypeptides involving azides is the Bergmann Degradation [1].
3. References
1. M. Bergmann, Science 79, 439 (1934)
Chemical ligation
Chemical ligation is a set of techniques used for creating long peptide or protein chains. It is the second step of a convergent approach. First smaller peptides containing 30-50 amino acids are prepared by conventional chemical peptide synthesis. They are then completely deprotected. Chemical ligation is the technique of selectively coupling these peptides in aqueous solution. With several coupling steps, proteins of up to 200-300 amino acids can be produced.
1. Methods of chemical ligation
There are various techniques described in literature. All of them involve a first kinetically controlled coupling step. The intermediate rearranges to form the amid bond. Older techniques rely on the presence of cysteine. Methods using removable auxiliary groups overcome this limitation. The most important one is Staudinger ligation.
1.1. Prior thiol capture
Prior thiol capture was the first ligation process. It was developed in the 1980s. Through a disulfide bridge N-terminal cysteine is brought into proximity of the C-terminal amino acid of the other peptide.
1.2. Native chemical ligation
Developed in the 1990s, native chemical ligation is a method commonly used. N-terminal cysteine reacts with a C-terminal thioester.
1.3. Expressed protein ligation
By exploiting naturally present inteins it is possible to prepare a recombinant C-terminal thioester. This removes the size restriction of the C-terminal thioester fragment. The recombinant thioester can be ligated to a synthetic peptide bearing an N-terminal cysteine. Native Chemical Ligations of this kind using recombinant C-terminal thioesters is known as Expressed protein ligation.
1.4. Acyl initiated capture
Acyl initiated capture is similar to native chemical ligation, except that peptide thioacid is brought into reaction with N-terminal bromo-alanine. The product is a peptide including cysteine.
1.5. Staudinger ligation
With Staudinger ligation, developed in 2000, peptide chains can be linked independently of the terminal amino acids. The method is based on the Staudinger reaction.
2. References
Nilsson BL, Soellner MB, Raines RT. 2005. Chemical Synthesis of Proteins. Annu. Rev. Biophys. Biomol. Struct. 34:91-118
Native chemical ligation
Native chemical ligation is a common form of chemical ligation, a technique for constructing a large peptide from two or more smaller peptides. In native chemical ligation a peptide containing a C-terminal thioester reacts with another peptide containing an N-terminal cysteine, in the presence of an exogenous thiol catalyst. In a thermodynamically-controlled, freely reversible first step a transthioesterification occurs. The product rearranges irreversibly under the usual reaction conditions to form the desired amide bond. The process was developed in the laboratory of Stephen Kent at The Scripps Research Institute in 1994.
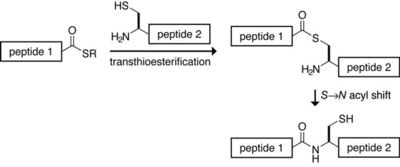
Native chemical ligation
Peptide-thioesters to be used in native chemical ligation are usually prepared by Boc chemistry SPPS; the thioester piece cannot be synthesized with a nucleophilic base, thus disfavoring Fmoc chemistry. Fmoc techniques for generating thioesters involving the modifications of the Kenner ‘safety catch’ linker are known. Protecting groups on the N-terminal piece cannot have constituents which release as aldehydes or ketones since these will cap the N-terminal cysteine. For the same reason, acetone should be avoided in general use, particularly prior to lyophilization and in washing glassware.
The payoff is that coupling long peptides by this technique is in many cases nearly quantitative and provides synthetic access to proteins otherwise impossible to make, due to length or decoration by posttranslational modification.
A limitation of this technique is that cysteine has to be part of the produced protein at a suitable place. For some proteins homocysteine can be used and methylated after coupling to form methionine. Cysteine can also be desulfurized to alanine. Alternately, ligation auxiliaries can be used that mimic an N-terminal cysteine for the ligation reaction, but which can be removed after synthesis.
Polypeptide C-terminal thioesters produced by recombinant DNA techniques can be reacted with an N-terminal Cys containing polypeptide by the same native ligation chemistry to provide very large semisynthesized proteins. Thus, by exploiting nature’s inteins to prepare a recombinant C-terminal thioester, the size restriction of the reacting peptide segment is removed. Similarly, a recombinant protein containing an N-terminal Cys can be reacted with a synthetic polypeptide thioester. Native Chemical Ligation of this kind using a recombinant polypeptide segment is known as Expressed Protein Ligation.
References
• Dawson PE, Muir TW, Clark-Lewis I, Kent, SBH. 1994. Synthesis of Proteins by Native Chemical Ligation. Science 266:776-779.
• Muir TW, Sondhi D, Cole PA. 1998. Expressed Protein Ligation: A General Method for Protein Engineering. Proc. Natl. Acad. Sci. USA 95:6705-6710.
• Nilsson BL, Soellner MB, Raines RT. 2005. Chemical Synthesis of Proteins. Annu. Rev. Biophys. Biomol. Struct. 34:91-118
Polyethylene glycol
Polyethylene glycol (PEG) and polyethylene oxide (PEO) are polymers composed of repeating subunits of identical structure, called monomers, and are the most commercially important polyethers. Poly (ethylene glycol) or poly (ethylene oxide) refers to an oligomer or polymer of ethylene oxide. The two names are chemically synonymous, but historically PEG has tended to refer to shorter polymers, PEO to longer.[1] PEG and PEO are liquids or low-melting solids, depending on their molecular weights. Both are prepared by polymerization of ethylene oxide. While PEG and PEO with different molecular weights find use in different applications and have different physical properties (e.g. viscosity) due to chain length effects, their chemical properties are nearly identical. Derivatives of PEG and PEO are in common use, the most common derivative being the methyl ether (methoxypoly (ethylene glycol)), abbreviated mPEG.
Polyethylene glycol
Their melting points vary depending on the Formula Weight of the polymer. PEG or PEO has the following structure:
HO-(CH2-CH2-O)n-H
The numbers that are often included in the names of PEGs and PEOs indicate their average molecular weights, e.g. a PEG with n=80 would have an average molecular weight of approximately 3500 daltons and would be labeled PEG 3500. Most PEGs and PEOs include molecules with a distribution of molecular weights, i.e. they are polydisperse. The size distribution can be characterized statistically by its weight average molecular weight (Mw) and its number average molecular weight (Mn), the ratio of which is called the polydispersity index (Mw/Mn). Mw and Mn can be measured by mass spectroscopy.
PEGylation is the act of covalently coupling a PEG structure to another larger molecule, for example, a therapeutic protein (which is then referred to as PEGylated). PEGylated interferon alfa-2a or -2b is a commonly used injectable treatment for Hepatitis C infection.
PEG is soluble in water, methanol, benzene, dichloromethane and is insoluble in diethyl ether and hexane. It is coupled to hydrophobic molecules to produce non-ionic surfactants.
1. Production
Poly (ethylene glycol) is produced by interaction of calculated amount of ethylene oxide with water, ethylene glycol or ethylene glycol oligomers.[2] The reaction is catalyzed by acidic or basic catalysts. Ethylene glycol and its oligomers are preferable as a starting material than water, because it allows the creation of polymers with narrow molecular weight distribution (low polydispersity). Polymer chain length depends on the ratio of reactants.
HOCH2CH2OH + n(CH2CH2O) → HO(CH2CH2O)n+1H
Depending on the catalyst type the mechanism of polymerization can be cationic or anionic. Anionic mechanism is more preferable because it allows one to obtain PEG with low polydispersity. Polymerization of ethylene oxide is an exothermic process. Contamination of ethylene oxide with catalysts such as alkalis or metal oxides or overheating can lead to runaway polymerization which can end with an explosion after few hours.
Polyethylene oxide or high-molecular polyethylene glycol is synthesized by suspension polymerization. It is necessary to hold the growing polymer chain in solution in the course of the polycondensation process. The reaction is catalyzed by magnesium-, aluminium- or calcium- organoelement compounds. To prevent coagulation of polymer chains from solution, chelating additives such as dimethylglyoxime are used.
Alkali catalysts such as sodium hydroxide NaOH, potassium hydroxide KOH or sodium carbonate Na2CO3 are used to prepare low-molecular polyethylene glycol.
2. Clinical uses
Polyethylene glycol has a low toxicity[2] and is used in a variety of products. It is the basis of a number of laxatives (e.g. macrogol-containing products such as Movicol and polyethylene glycol 3350, or MiraLax or GlycoLax). It is the basis of many skin creams, as cetomacrogol, and sexual lubricants, frequently combined with glycerin. Whole bowel irrigation (polyethylene glycol with added electrolytes) is used for bowel preparation before surgery or colonoscopy and drug overdoses. It is sold under the brand names GoLYTELY, GlycoLax, Fortrans, TriLyte, and Colyte. When attached to various protein medications, polyethylene glycol allows a slowed clearance of the carried protein from the blood. This makes for a longer acting medicinal effect and reduces toxicity, and it allows longer dosing intervals. Examples include PEG-interferon alpha which is used to treat hepatitis C and PEG-filgrastim (Neulasta®) which is used to treat neutropenia. It has been shown that polyethylene glycol can improve healing of spinal injuries in dogs.[3] One of the earlier findings that polyethylene glycol can aid in nerve repair came from the University of Texas (Krause and Bittner).[4] Polyethylene glycol is commonly used to fuse B-cells with myeloma cells in monoclonal antibody production.
3. Research for New Clinical Uses
• High-molecular weight PEG, e.g., PEG 8000, is a strikingly potent dietary preventive agent against colorectal cancer in animal models.[5] The Chemoprevention Database shows it is the most effective agent to suppress chemical carcinogenesis in rats. Cancer prevention in humans has not yet been tested in clinical trials.
• The injection of PEG 2000 into the bloodstream of guinea pigs after spinal cord injury leads to rapid recovery through molecular repair of nerve membranes.[6] The effect of this treatment to prevent paraplegia in humans after an accident is not known yet.
• Research is being done in the use of PEG to mask antigens on red blood cells. Various research institutes have reported that using PEG can mask antigens without damaging the functions and shape of the cell.
4. Other uses
PEG is used in a number of toothpastes as a dispersant; it binds water and helps keep gum uniform throughout the toothpaste. It is also under investigation for use in body armor[7] and tattoos to monitor diabetes.[8] Functional groups of PEG give polyurethane elastomers their “rubberiness”, for applications such as foams (foam rubber) and fibers (spandex). Its backbone structure is analogous to that of silicone, another elastomer.
Since PEG is a flexible, water-soluble polymer, it can be used to create very high osmotic pressures (tens of atmospheres). It also is unlikely to have specific interactions with biological chemicals. These properties make PEG one of the most useful molecules for applying osmotic pressure in biochemistry experiments, particularly when using the osmotic stress technique.
PEO (poly (ethylene oxide)) can serve as the separator and electrolyte solvent in lithium polymer cells. Its low diffusivity often requires high temperatures of operation, but its high viscosity even near its melting point allows very thin electrolyte layers. While crystallization of the polymer can degrade performance, many of the salts used to carry charge can also serve as a kinetic barrier to the formation of crystals. Such batteries carry greater energy for their weight than other lithium ion battery technologies.
When working with phenol in a laboratory situation, PEG 300 can be used on phenol skin burns to deactivate any residual phenol.
Poly (ethylene glycol) is also commonly used as a polar stationary phase for gas chromatography, as well as a heat transfer fluid in electronic testers.
PEG is included in many or all formulations of the soft drink Dr Pepper, purportedly as an anti-foaming agent.
PEG has also been used to preserve objects which have been salvaged from underwater, as was the case with the warship Vasa in Stockholm.[9] It replaces water in wooden objects, which makes the wood dimensionally stable and prevents warping or shrinking of the wood.
PEG is often seen (as a side effect) in mass spectrometry experiments with characteristic fragmentation patterns.
In the field of microbiology, PEG precipitation is used to concentrate viruses.
PEG is also used in lubricant eye drops. PEG derivatives such as narrow range ethoxylates are used as surfactants.
5. References
1. For example, in the online catalog[1] of Scientific Polymer Products, Inc., poly(ethylene glycol) molecular weights run up to about 20,000, while those of poly(ethylene oxide) have 6 or 7 digits.
2. Victor O. Sheftel (2000). Indirect Food Additives and Polymers: Migration and Toxicology. CRC, 1114-1116.
3. Lee Bowman. “Study on dogs yields hope in human paralysis treatment”, seattlepi.com, 4 December 2004.
4. T. L. Krause and G. D. Bittner (1990). “Rapid Morphological Fusion of Severed Myelinated Axons by Polyethylene Glycol”. PNAS 87 (4): 1471-1475.
5. D. E. Corpet, G. Parnaud, M. Delverdier, G. Peiffer and S. Tache (2000). “Consistent and Fast Inhibition of Colon Carcinogenesis by Polyethylene Glycol in Mice and Rats Given Various Carcinogens”. Cancer Res 60 (12): 3160-3164.
6. R. B. Borgens and D. Bohnert (2001). “Rapid recovery from spinal cord injury after subcutaneously administered polyethylene glycol”. Journal of Neuroscience Research 66 (6): 1179-1186. DOI:10.1002/jnr.1254.
7. Tonya Johnson. “Army Scientists, Engineers develop Liquid Body Armor”, Military.com, 21 April 2004.
8. “Tattoo to monitor diabetes”, BBC News, 1 September 2002.
9. Lars-Åke Kvarning, Bengt Ohrelius (1998), The Vasa – The Royal Ship, ISBN 91-7486-581-1, pp. 133-141
PEGylation
1. History
In 1970s, pioneering research by Davis, Abuchowski and colleagues foresaw the potential of the conjugation of Polyethylene glycol (PEG) to Proteins. This technique is now well established and is known as PEGylation.
PEGylation, is a process of attaching the strands of the polymer PEG to molecules most typically Peptides, Proteins, and Antibody fragments, that can help to meet the challenges of improving the safety and efficiency of many therapeutics. It produces alterations in the physiochemical properties including changes in conformation, electrostatic binding, hydrophobicity etc. These physical and chemical changes increase systemic retention of the therapeutic agent. Also, it can influence the binding affinity of the therapeutic moiety to the cell receptors and can alter the absorption and distribution patterns.
PEGylation, by increasing the molecular weight of a molecule, can impart several significant pharmacological advantages over the unmodified form, such as:
• Improved drug solubility
• Reduced dosage frequency, without diminished efficacy with potentially reduced toxicity
• Extended circulating life
• Increased drug stability
• Enhanced protection from proteolytic degradation
The PEGylated drugs are having the following commercial advantages also:
• Opportunities for new delivery formats and dosing regimens
• Extended patent life of previously approved drugs
2. PEGylated Pharmaceuticals on the Market
The clinical value of PEGylation is now well established. ADAGEN (PEG- bovine adenosine deaminase) manufactured by Enzon Pharmaceuticals, Inc., US was the first PEGylated protein approved by FDA in March 1990, to enter the market. It is used to treat X-linked severe combined immunogenicity syndrome, as an alternative to bone marrow transplantation and enzyme replacement by gene therapy. Since the introduction of ADAGEN, a large number of PEGylated protein and peptide pharmaceuticals have followed and many others are under clinical trial or under development stages. Some of the successful examples are:
• PEGasys: PEGylated alpha-interferons for use in the treatment of hepatis C (Hoffman-La Roche)
• PEG-Intron: PEGylated alpha -interferons for chronic hepatis C (Schering-Plough / Enzon)
• Oncaspar: PEGylated L-asparaginase for the treatment of acute lymphoblastic leukemia in patients who are hypersen-sitive to the native unmodified form of L-asparaginase (Enzon)
• Neulasta: PEGylated recombinant methionyl human granulocyte colony stimulating factor for severe cancer chemoth-erapy induced neutropenia (Amgen)
• Doxil: PEGylated liposome containing doxorubicin for the treatment of Cancer (Sequus)
3. PEG Moiety Properties
PEG is a particularly attractive polymer for conjugation. The specific characteristics of PEG moieties relevant to pharmaceutical applications are:
• Water solubility
• High mobility in solution
• Lack of toxicity and immunogenicity
• Ready clearance from the body
• Altered distribution in the body
4. PEGylation Process
The first step of the PEGylation is the suitable functionalization of the PEG polymer at one or both terminals. PEGs that are activated at each terminus with the same reactive moiety is known as “homobifunctional”, where as if the functional groups present are different, then the PEG derivative is referred as “heterobifunctional” or “heterofunctional.” The chemically active or activated derivatives of the PEG polymer are prepared to attach the PEG to the desired molecule.
The choice of the suitable functional group for the PEG derivative is based on the type of available reactive group on the molecule that will be coupled to the PEG. For proteins, typical reactive amino acids include lysine, cysteine, histidine, arginine, aspartic acid, glutamic acid, serine, threonine, tyrosine. The N-terminal amino group and the C-terminal carboxylic acid can also be used.
The techniques used to form first generation PEG derivatives are generally reacting the PEG polymer with a group that is reactive with hydroxyl groups, typically anhydrides, acid chlorides, chloroformates and carbonates. In the second generation PEGylation chemistry more efficient functional groups such as aldehyde, esters, amides etc made available for conjugation.
As applications of PEGylation have become more and more advanced and sophisticated, there has been an increase in need for heterobifunctional PEGs for conjugation. These heterobifunctional PEGs are very much useful in linking two entities, where a hydrophilic, flexible and biocompatible spacer is needed. Preferred end groups for heterobifunctional PEGs are maleimide, vinyl sulphones, pyridyl disulphide, amine, carboxylic acids and NHS esters.
5. References
• Abuchowski, McCoy, Palczuk, van Es and Davis (1977). “Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase.” Journal of Biological Chemistry 252(11): 3582-3586.
• Fee (2003). “Size-exclusion reaction chromatography (SERC): A new technique for protein PEGylation.” Biotechnology and Bioengineering 82(2): 200-206.
• Fee and Alstine (2006). “PEG-proteins: Reaction engineering and separation issues.” Chemical Engineering Science 61(3): 924-939.
• Kodera, Matsushima, Hiroto, Nishimura, Ishii, Ueno and Inada (1998). “Pegylation of proteins and bioactive substances for medical and technical applications.” Progress in Polymer Science 23(7): 1233-1271.
• Morar, Jeffrey and Mark (2006). “PEGylation of Proteins: A Structural Approach.” Biopharm International 19(4): 34.
• Roberts, Bentley and Harris (2002). “Chemistry for peptide and protein PEGylation.” Advanced Drug Delivery Reviews 54(4): 459-476.
• Veronese (2001). “Peptide and protein PEGylation: a review of problems and solutions.” Biomaterials 22(5): 405-417.
• Veronese and Harris (2002). “Introduction and overview of peptide and protein pegylation.” Advanced Drug Delivery Reviews 54(4): 453-456.
• Veronese and Pasut (2005). “PEGylation, successful approach to drug delivery.” Drug Discovery Today 10(21): 1451-1458.
Freeze drying
Freeze drying (also known as lyophilization) is a dehydration process typically used to preserve a perishable material or make the material more convenient for transport. Freeze drying works by freezing the material and then reducing the surrounding pressure and adding enough heat to allow the frozen water in the material to sublime directly from the solid phase to gas.
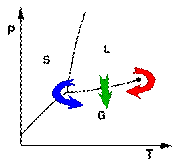
In a typical phase diagram, the boundary between gas and liquid runs from the triple point to the critical point. Freeze drying (blue arrow) brings the system around the triple point, avoiding the direct liquid-gas transition seen in ordinary drying (green arrow).
Contents
1. Freeze drying protectants
2. The Freeze-drying process
2.1. Freezing
2.2. Primary Drying
2.3. Secondary Drying
3. Properties of Freeze-dried Products
4. Uses of Freeze-drying
4.1. Pharmaceutical and Bio-Tech
4.2. Food Industry
4.3. Technological Industry
4.4. Other Uses
5. Freeze-drying Equipment
6. References
1. Freeze drying protectants
Similar to cryoprotectants, some molecules protect freeze dried material. Known as lyoprotectants, these molecules are typically polyhydroxy compounds such as sugars (mono-, di-, and polysaccharides), polyalcohols, and their derivatives. Trehalose and sucrose are natural lyoprotectants. Trehalose is produced by a variety of plant, fungi, and invertebrate animals that remain in a state of suspended animation during periods of drought (also known as anhydrobiosis).
2. The Freeze-drying process
There are three stages in the complete freeze-drying process: Freezing, Primary Drying, and Secondary Drying.
2.1. Freezing
The freezing process consists of freezing the material. In a lab, this is often done by placing the material in a freeze-drying flask and rotating the flask in a bath, called a shell freezer, which is cooled by mechanical refrigeration, dry ice and methanol, or liquid nitrogen. On a larger-scale, freezing is usually done using a freeze-drying machine. In this step, it is important to freeze the material at a temperature below the eutectic point of the material. Since the eutectic point occurs at the lowest temperature where the solid and liquid phase of the material can coexist, freezing the material at a temperature below this point ensures that sublimation rather than melting will occur in the following steps. Larger crystals are easier to freeze dry. To produce larger crystals the product should be frozen slowly or can be cycled up and down in temperature. This cycling process is called annealing.
Amorphous (glassy) materials do not have a eutectic point, but do have a critical temperature, below which the product must be maintained to prevent melt-back or collapse during primary and secondary drying.
2.2. Primary Drying
During the primary drying phase the pressure is lowered and enough heat is supplied to the material for the water to sublimate. The amount of heat necessary can be calculated using the sublimating molecules’ latent heat of sublimation. In this initial drying phase about 98% of the water in the material is sublimated. This phase may be slow, because if too much heat is added the material’s structure could be altered.
In this phase, pressure is controlled through the application of partial vacuum. The vacuum speeds sublimation making it useful as a deliberate drying process. Furthermore, a cold condenser chamber and/or condenser plates provide a surface(s) for the water vapour to re-solidify on. This condenser plays no role in keeping the material frozen; rather, it prevents water vapor from reaching the vacuum pump, which could degrade the pump’s performance. Condenser temperatures are typically below −50 °C (-58°F).
2.3. Secondary Drying
The secondary drying phase aims to sublimate the water molecules that are adsorbed during the freezing process, since the mobile water molecules were sublimated in the primary drying phase. This part of the freeze-drying process is governed by the material’s adsorption isotherms. In this phase, the temperature is raised even higher than in the primary drying phase to break any physico-chemical interactions that have formed between the water molecules and the frozen material. Usually the pressure is also lowered in this stage to encourage sublimation. However, there are products that benefit from increased pressure as well.
After the freeze drying process is complete, the vacuum is usually broken with an inert gas, such as nitrogen, before the material is sealed.
3. Properties of Freeze-dried Products
If a freeze-dried substance is sealed to prevent the reabsorption of moisture, the substance may be stored at room temperature without refrigeration, and be protected against spoilage for many years. Preservation is possible because the greatly reduced water content inhibits the action of microorganisms and enzymes that would normally spoil or degrade the substance.
Freeze drying also causes less damage to the substance than other dehydration methods using higher temperatures. Freeze drying does not usually cause shrinkage or toughening of the material being dried. In addition, flavours and smells generally remain unchanged, making the process popular for preserving food. Unfortunately, water is not the only chemical capable of sublimation and the loss of other volatile compounds such as acetic acid (vinegar) and alcohols can yield undesirable results.
Freeze-dried products can be rehydrated (reconstituted) much more quickly and easily because it leaves microscopic pores. The pores are created by the ice crystals that sublimate, leaving gaps or pores in its place. This is especially important when it comes to pharmaceutical uses. Lyophilization can also be used to increase the shelf life of some pharmaceuticals for many years.
4. Uses of Freeze-drying
4.1. Pharmaceutical and Bio-Tech
Pharmaceutical companies often use freeze drying to increase the shelf life of products, such as vaccines and other injectables. By removing the water from the material and sealing the material in a vial, the material can be easily stored, shipped and later reconstituted to its original form for injection.
4.2. Food Industry
The process has been popularized in the forms of freeze-dried ice cream, an example of astronaut food. It is also popular and convenient for hikers because the reduced weight allows them to carry more food and reconstitute it with available water. Instant coffee is sometimes freeze dried, despite high costs of freeze dryers. The coffee is often dried by vaporization in a hot air flow, or by projection on hot metallic plates. Currently, the freeze-drying process is used more commonly in the pharmaceutical industry.
4.3. Technological Industry
In chemical synthesis, products are often lyophilized to make them more stable, or easier to dissolve in water for subsequent use.
In bioseparations, freeze drying can also be used as a late-stage purification procedure, because it can effectively remove solvents. Furthermore, it is capable of concentrating substances with low molecular weights that are too small to be removed by a filtration membrane.
Freeze-drying is a relatively expensive process. The equipment is about three times as expensive as the equipment used for other separation processes, and the high energy demands lead to high energy costs. Furthermore, freeze drying also has a long process time, because the addition of too much heat to the material can cause melting or structural deformations. Therefore, freeze drying is often reserved for materials that are heat-sensitive, such as proteins, enzymes, microorganisms, and blood plasma. The low operating temperature of the process leads to minimal damage of these heat-sensitive products.
4.4 Other Uses
Recently, some taxidermists have begun using freeze drying to preserve animals.
Organizations such as the Document Conservation Laboratory at the United States National Archives and Records Administration (NARA) have done studies on freeze drying as a recovery method of water-damaged books and documents. While recovery is possible, restoration quality depends on the material of the documents. If a document is made of a variety of materials, which have different absorption properties, expansion will occur at a non-uniform rate which could lead to deformations. Water can also cause mold to grow or make inks bleed. In these cases, freeze drying may not be an effective restoration method.
In high altitude environments, the low temperatures and pressures can sometimes produce natural mummies by a process of freeze-drying.
5. Freeze-drying Equipment
There are essentially three categories of freeze dryers: rotary evaporators, manifold freeze dryers, and tray freeze dryers.
Rotary freeze dryers are usually used with liquid products, such as pharmaceutical solutions and tissue extracts.
Manifold freeze dryers are usually used when drying a large amount of small containers and the product will be used in a short period of time. A manifold dryer will dry the product to less than 5% moisture content. Without heat only primary drying (removal of the unbound water) can be achieved. A heater needs to be added for secondary drying, which will remove the bound water and will produce a lower moisture content.
Tray freeze dryers are more sophisticated and are used to dry a variety of materials. A tray freeze dryer is used to produce the driest product for long-term storage. A tray freeze dryer allows the product to be frozen in place and performs both primary (unbound water removal) and secondary (bound water removal) freeze drying, thus producing the driest possible end-product. Tray freeze dryers can dry product in bulk or in vials. When drying in vials, the freeze dryer is supplied with a stoppering mechanism that allows a stopper to be pressed into place sealing the vial before it is exposed to the atmosphere. This is used for long term storage, such as vaccines.
6. References
• Harris, E. L. V. and S. Angal (1989). Protein Purification Methods. Oxford University Press. ISBN 0-19-963003-8
• Kennedy, John F. and Joaquim M. S. Cabral (1993). Recovery Processes for Biological Materials. John Wiley & Sons Ltd.
• http://www.archives.gov/preservation/conservation/drying-methods-01.html
