
Peptide
This entry is from Wikipedia, the leading user-contributed encyclopedia.
• N-terminal end
• C-terminal end
• Disulfide bond
• Peptide bond
• Peptide library
• Peptide sequence
• Peptide sequence tag
• Peptide mass fingerprinting
• Antimicrobial peptides
• Beta-peptide
• Cyclotides
• Glycopeptide antibiotic
• Nonribosomal peptide
• Signal peptide
• Microcystin
• Peptide nucleic acid
N-terminal end
The N-terminus (also known as the amino-terminus, NH2-terminus, N-terminal end or amine-terminus) refers to the end of a protein or polypeptide terminated by an amino acid with a free amine group (-NH2). The convention for writing peptide sequences is to put the N-terminus on the left and write the sequence from N- to C-terminus.
1. Chemistry
Each amino acid has a carboxyl group and an amine group, and amino acids link to one another to form a chain by a dehydration reaction by joining the amine group of one amino acid to the carboxyl group of the next. Thus polypeptide chains have an end with an unbound carboxyl group, the C-terminus, and an end with an amine group, the N-terminus.
When the protein is translated from messenger RNA, it is created from N-terminus to C-terminus. The amino end of an amino acid (on a charged tRNA) during the elongation stage of translation, attaches to the carboxyl end of the growing or nascent chain. Since the start codon of the genetic code codes for the amino acid methionine, most protein sequences start with a methionine (more specifically: the modified version N-formylmethionine, fMet). However, some proteins are modified posttranslationally, for example by cleavage from a protein precursor, and therefore may have different amino acids at their N-terminus.
2. Function
2.1. N-terminal targeting signals
The N-terminus is the first part of the protein that exits the ribosome during protein biosynthesis. It often contains sequences that act as targeting signals, basically intracellular zip codes, that allow for the protein to be delivered to its designated location within the cell. The targeting signal is usually cleaved off after successful targeting by a processing peptidase.
2.1.1. Signal peptide
The N-terminal signal peptide is recognized by the signal recognition particle (SRP) and results in the targeting of the protein to the secretory pathway. In eukaryotic cells, these proteins are synthesized at the rough endoplasmic reticulum. In prokaryotic cells, the proteins are exported across the cell membrane. In chloroplasts, signal peptides target proteins to the thylakoids.
2.1.2. Mitochondrial targeting peptide
The N-terminal mitochondrial targeting peptide (mtTP) allows for the protein to be imported into the mitochondrion.
2.1.3. Chloroplast targeting peptide
The N-terminal chloroplast targeting peptide (cpTP) allows for the protein to be imported into the chloroplast.
2.2. N-terminal modifications
Some proteins are modified posttranslationally by the addition of membrane anchors that allow the protein to associate with membrane without having a transmembrane domain. The N-terminus (as well as the C-terminus) of a protein can be modified this way.
2.2.1. N-Myristoylation
Main article: Myristoylation
The N-terminus can be modified by the addition of a myristoyl anchor. Proteins that are modified this way contain a consensus motif at their N-terminus as a modification signal.
2.2.2. N-Acylation
The N-terminus can also be modified by the addition of a fatty acid anchor to form N-acylated proteins. The most common form of such modification is the addition of a palmitoyl group.
C-terminal end
The C-terminus (also known as the carboxyl-terminus, carboxy-terminus, C-terminal end, or COOH-terminus) of a protein or polypeptide is the end of the amino acid chain terminated by a free carboxyl group (-COOH). The convention for writing peptide sequences is to put the C-terminal end on the right and write the sequence from N- to C-terminus.
1. Chemistry
Each amino acid has a carboxyl group and an amine group, and amino acids link to one another to form a chain by a dehydration reaction by joining the amine group of one amino acid to the carboxyl group of the next. Thus polypeptide chains have an end with an unbound carboxyl group, the C-terminus, and an end with an amine group, the N-terminus. Proteins are synthesized starting from the N-terminus and ending at the C-terminus.
2. Function
2.1. C-terminal retention signals
While the N-terminus of a protein often contains targeting signals, the C-terminus can contain retention signals for protein sorting. The most common ER retention signal is the amino acid sequence -KDEL (or -HDEL) at the C-terminus, which keeps the protein in the endoplasmic reticulum and prevents it from entering the secretory pathway.
2.2. C-terminal modifications
The C-terminus of proteins can be modified posttranslationally, most commonly by the addition of a lipid anchor to the C-terminus that allows the protein to be inserted into a membrane without having a transmembrane domain.
2.2.1. Prenylation
One form of C-terminal modification is prenylation. During prenylation, a farnesyl- or geranylgeranyl-isoprenoid membrane anchor is added to a cysteine residue near the C-terminus. Small, membrane-bound G proteins are often modified this way.
2.2.2. GPI anchors
Another form of C-terminal modification is the addition of a phosphoglycan, glycosylphosphatidylinositol (GPI), as a membrane anchor. The GPI anchor is attached to the C-terminus after proteolytic cleavage of a C-terminal propeptide. The most prominent example for this type of modification is the prion protein.
2.3. C-terminal domain
The C-terminal domain (CTD) of some proteins has specialized functions.
2.3.1. CTD of RNA polymerase
The carboxy-terminal domain of RNA polymerase II typically consists of up to 52 repeats of the sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser [1]. Other proteins often bind the C-terminal domain of RNA polymerase in order to activate polymerase activity. It is the protein domain which is involved in the initiation of DNA transcription, the capping of the RNA transcript, and attachment to the spliceosome for RNA splicing.
Disulfide bond
In chemistry, a disulfide bond is a single covalent bond derived from the coupling of thiol groups. The linkage is also called an SS-bond or disulfide bridge. The overall connectivity is therefore C-S-S-C. The terminology is almost exclusively used in biochemistry, bioinorganic and bioorganic chemistry. Formally the connection is called a persulfide, in analogy to a peroxide (R-O-O-R), but this terminology is rare.
Three sulfur atoms singly bonded in a sequence are sometimes called a trisulfide bond, although there are in fact two S-S bonds. Disulfide bonds are usually formed from the oxidation of sulfhydryl (-SH) groups, as depicted formally in Figure 1.
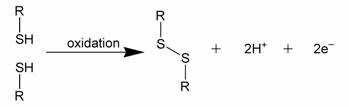
Figure 1: Formal depiction of disulfide bond formation as an oxidation.
1. Disulfide bonds in proteins
Disulfide bonds play an important role in the folding and stability of some proteins, usually proteins secreted to the extracellular medium. Since most cellular compartments are a reducing environment, disulfide bonds are generally unstable in the cytosol (with some exceptions noted below).
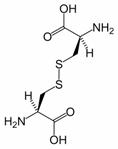
Figure 2: Cystine is composed of two cysteines linked by a disulfide bond (shown here in its neutral form).
Disulfide bonds in proteins are formed between the thiol groups of cysteine residues. The other sulfur-containing amino acid, methionine, cannot form disulfide bonds. A disulfide bond is typically denoted by hyphenating the abbreviations for cysteine, e.g., the “Cys26-Cys84 disulfide bond”, or the “26-84 disulfide bond”, or most simply as “C26-C84” where the disulfide bond is understood and does not need to be mentioned. The prototype of a protein disulfide bond is the two-amino-acid peptide, cystine, which is composed of two cysteine amino acids joined by a disulfide bond (shown in Figure 2 in its unionized form). The structure of a disulfide bond can be described by its Xss dihedral angle between the Cβ − Sγ − Sγ − Cβ atoms, which is usually close to ±90°.
The disulfide bond stabilizes the folded form of a protein in several ways: 1) It holds two portions of the protein together, biasing the protein towards the folded topology. Expressed differently, the disulfide bond destabilizes the unfolded form of the protein by lowering its entropy. 2) The disulfide bond may form the nucleus of a hydrophobic core of the folded protein, i.e., local hydrophobic residues may condense around the disulfide bond and onto each other through hydrophobic interactions. 3) Related to #1 and #2, the disulfide bond link two segments of the protein chain, the disulfide bond increases the effective local concentration of protein residues and lowers the effective local concentration of water molecules. Since water molecules attack amide-amide hydrogen bonds and break up secondary structure, a disulfide bond stabilizes secondary structure in its vicinity. For example, researchers have identified several pairs of peptides that are unstructured in isolation, but adopt stable secondary and tertiary structure upon forming a disulfide bond between them.
Disulfide bonds in proteins are formed by thiol-disulfide exchange reactions. A disulfide species is a particular pairing of cysteines in a disulfide-bonded protein and is usually depicted by listing the disulfide bonds in parentheses, e.g., the “(26-84, 58-110) disulfide species”. A disulfide ensemble is a grouping of all disulfide species with the same number of disulfide bonds, and is usually denoted as the 1S ensemble, the 2S ensemble, etc. for disulfide species having one, two, etc. disulfide bonds. Thus, the (26-84) disulfide species belongs to the 1S ensemble, whereas the (26-84, 58-110) species belongs to the 2S ensemble. The single species with no disulfide bonds is usually denoted as R for “fully reduced”. Under typical conditions, disulfide reshuffling is much faster than the formation of new disulfide bonds or their reduction; hence, the disulfide species within an ensemble equilibrate more quickly than between ensembles.
The native form of a protein is usually a single disulfide species, although some proteins may cycle between a few disulfide states as part of their function, e.g., thioredoxin. In proteins with more than two cysteines, non-native disulfide species may be formed, which are almost always unfolded. As the number of cysteines increases, the number of nonnative species increases factorially. The number of ways of forming p disulfide bonds from n cysteine residues is given by the formula
For example, an eight-cysteine protein such as ribonuclease A has 105 different four-disulfide species, only one of which is the native disulfide species. Isomerases have been identified that catalyze the interconversion of disulfide species, accelerating the formation of the native disulfide species.
Disulfide species that have only native disulfide bonds (but not all of them) are denoted by des followed by the lacking native disulfide bond(s) in square brackets. For example, the des[40-95] disulfide species has all the native disulfide bonds except that between cysteines 40 and 95. Disulfide species that lack one native disulfide bond are frequently folded, particularly if the missing disulfide bond is exposed to solvent in the folded, native protein.
2. In prokaryotes
Disulfide bonds play an important protective role for bacteria as a reversible switch that turns a protein on or off when bacterial cells are exposed to oxidation reactions. Hydrogen peroxide (H2O2) in particular could severely damage DNA and kill the bacterium at low concentrations if it weren’t for the protective action of the SS-bond.
3. In rubber
Disulfide bonds also play a significant role in the vulcanization of rubber.
4. In eukaryotes
In eukaryotic cells, disulfide bonds are generally formed in the lumen of the RER (rough endoplasmic reticulum) but not in the cytosol. This is due to the oxidative environment of the ER and the reducing environment of the cytosol (see glutathione). Thus disulfide bonds are mostly found in secretory proteins, lysosomal proteins, and the exoplasmic domains of membrane proteins.
There are notable exceptions to this rule. A number of cytosolic proteins have cysteine residues in proximity to each other that function as oxidation sensors; when the reductive potential of the cell fails, they oxidize and trigger cellular response mechanisms. Vaccinia virus also produces cytosolic proteins and peptides that have many disulfide bonds; although the reason for this is unknown presumably they have protective effects against intracellular proteolysis machinery.
Disulfide bonds are also formed within and between protamines in the sperm chromatin of many mammalian species.
5. In hair and feathers
Hair is a biological polymer, with over 90% of its dry weight made of proteins called keratins. Under normal conditions, human hair contains around 10% water, which modifies its mechanical properties considerably. Hair proteins are held together by disulfide bonds, from the amino acid cysteine. These links are very robust: for example, virtually intact hair has been recovered from ancient Egyptian tombs, and the disulfide links also cause hair (and feathers which have similar keratins) to be extremely resistant to protein digestive enzymes. Different parts of the hair and feather have different cysteine levels, leading to harder or softer material.
Breaking and making disulfide bonds governs the phenomenon of wavy or frizzy hair. It is breaking and remaking of the disulfide bonds which is the basis for the permanent wave in hairstyling.
In feathers, the high disulfide content dictates the high sulfur content of bird eggs, which need to contain enough sulfur to feather the chick.
In both hair and feathers, the high sulfur content due to the high number of disulfides causes the disagreeable smell of the material when it is burned.
6. In organic chemistry
The Zincke disulfide cleavage is a classic organic reaction in which a disulfide is converted to a sulfur halide R-S-X with X = Br, Cl by reaction with bromine or chlorine.[1][2][3][4]
7. General references
• Sela M and Lifson S. (1959) “On the Reformation of Disulfide Bridges in Proteins”, Biochimica et Biophysica Acta, 36, 471-478.
• Stark GR. (1977) “Cleavage at cysteine after cyanylation”, Methods in Enzymology, 11, 238-255.
• Thornton JM. (1981) “Disulphide Bridges in Globular Proteins”, Journal of Molecular Biology, 151, 261-287.
• Thannhauser TW, Konishi Y and Scheraga HA. (1984) “Sensitive Quantitative Analysis of Disulfide Bonds in Polypeptides and Proteins”, Analytical Biochemistry, 138, 181-188.
• Wu J and Watson JT. (1998) “Optimization of the Cleavage Reaction for Cyanylated Cysteinyl Proteins for Eficient and Simplified Mas Mapping”, Analytical Biochemistry, 258, 268-276.
• Futami J, Tada H, Seno M, Ishikami S and Yamada H. (2000) “Stabilization of Human RNase 1 by Introduction of a Disulfide Bond between Residues 4 and 118”, J. Biochem., 128, 245-250.
8. References
1. Zincke, Ber. 44, 770 (1911); Zincke and Farr, Ann. 391, 63 (1912)
2. The conversion of di-o-nitrophenyl disulfide to o-nitrophenylsulfur chloride Organic Syntheses, Coll. Vol. 2, p.455 (1943); Vol. 15, p.45 (1935) Link
3. Related reactions : Organic Syntheses, Coll. Vol. 9, p.662 (1998); Vol. 74, p.124 (1997) Link
4. Organic Syntheses, Coll. Vol. 5, p.709 (1973); Vol. 40, p.62 (1960) Link
9. External links
• Synthesis of Disulfides
• Disulfide bonds and hair
• Protein disulfide bond formation in prokaryotes
• Oxidative protein folding in eukaryotes : mechanisms and consequences
• The human protein disulfide isomerase family: substrate interactions and functional properties
Peptide bond
A peptide bond is a chemical bond formed between two molecules when the carboxyl group of one molecule reacts with the amino group of the other molecule, releasing a molecule of water (H2O). This is a dehydration synthesis reaction (also known as a condensation reaction), and usually occurs between amino acids. The resulting CO-NH bond is called a peptide bond, and the resulting molecule is an amide. The four-atom functional group -C(=O)NH- is called an amide group or (in the context of proteins) a peptide group. Polypeptides and proteins are chains of amino acids held together by peptide bonds, as is the backbone of PNA. Polyamides, such as nylons and aramids, are synthetic molecules (polymers) that possess peptide bonds.

Figure 1: Dehydration synthesis (condensation) reaction forming an amide
A peptide bond can be broken by amide hydrolysis (the adding of water). The peptide bonds in proteins are metastable, meaning that in the presence of water they will break spontaneously, releasing about 10 kJ/mol of free energy, but this process is extremely slow. In living organisms, the process is facilitated by enzymes. Living organisms also employ enzymes to form peptide bonds; this process requires free energy. The wavelength of absorbance for a peptide bond is 190-230nm.
Contents
1. Resonance forms of the peptide group
2. Cis/trans isomers of the peptide group
3. Chemical reactions
4. References
1. Resonance forms of the peptide group
The amide group has two resonance forms, which confer several important properties. First, it stabilizes the group by roughly 20 kcal/mol, making it less reactive than many similar groups (such as esters). The resonance suggests that the amide group has a partial double bond character, estimated at 40% under typical conditions. The peptide group is uncharged at all normal pH values, but its double-bonded resonance form gives it a unusually large dipole moment, roughly 3.5 Debye (0.7 electron-angstrom). These dipole moments can line up in certain secondary structures (such as the α-helix), producing a large net dipole.
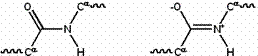
Figure 2: Resonance forms of a typical peptide group. The uncharged, single-bonded form (typically ~60%) is shown on the left, whereas the charged, double-bonded form (typically ~40%) is on the right.
The partial double bond character can be strengthened or weakened by modifications that favor one resonance form over another. For example, the double-bonded form is disfavored in hydrophobic environments, because of its charge. Conversely, donating a hydrogen bond to the amide oxygen or accepting a hydrogen bond from the amide nitrogen should favor the double-bonded form, because the hydrogen bond should be stronger to the charged form than to the uncharged, single-bonded form. By contrast, donating a hydrogen bond to an amide nitrogen in an X-Pro peptide bond should favor the single-bonded form; donating it to the double-bonded form would give the nitrogen five quasi-covalent bonds! (See Figure 3.)
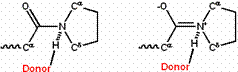
Figure 3: Donation of an H-bond to an X-Pro peptide group favors the single-bonded resonance form (left) over the double-bonded form (right).
Similarly, a strongly electronegative substituent (such as fluorine) near the amide nitrogen favors the single-bonded form, by competing with the amide oxygen to “steal” an electron from the amide nitrogen (See Figure 4.)
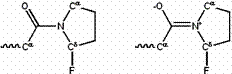
Figure 4: An electronegative substituent near the amide nitrogen favors the single-bonded resonance form (left) over the double-bonded form (right).
2. Cis/trans isomers of the peptide group
The partial double bond renders the amide group planar, occurring in either the cis or trans isomers. In the unfolded state of proteins, the peptide groups are free to isomerize and adopt both isomers; however, in the folded state, only a single isomer is adopted at each position (with rare exceptions). The trans form is preferred overwhelmingly in most peptide bonds (roughly 1000:1 ratio in trans:cis populations). However, X-Pro peptide groups tend to have a roughly 3:1 ratio, presumably because the symmetry between the Cα and Cδ atoms of proline makes the cis and trans isomers nearly equal in energy (See figure, below).
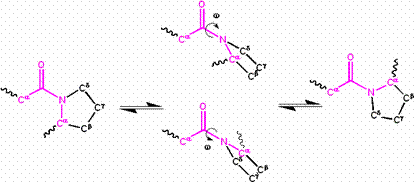
Isomerization of an X-Pro peptide bond. Cis and trans isomers are at far left and far right, respectively, separated by the transition states.
The dihedral angle associated with the peptide group (defined by the four atoms ) is denoted ω; for the cis isomer and for the trans isomer. Amide groups can isomerize about the C-N bond between the cis and trans forms, albeit slowly (20 seconds at room temperature). The transition states requires that the partial double bond be broken, so that the activation energy is roughly 20 kcal/mol (See Figure below). However, the activation energy can be lowered (and the isomerization catalyzed) by changes that favor the single-bonded form, such as placing the peptide group in a hydrophobic environment or donating a hydrogen bond to the nitrogen atom of an X-Pro peptide group. Both of these mechanisms for lowering the activation energy have been observed in peptidyl prolyl isomerases (PPIases), which are naturally occurring enzymes that catalyze the cis-trans isomerization of X-Pro peptide bonds.
Conformational protein folding is usually much faster (typically 10-100 ms) than cis-trans isomerization (10-100 s). A nonnative isomer of some peptide groups can disrupt the conformational folding significantly, either slowing it or preventing it from even occurring until the native isomer is reached. However, not all peptide groups have the same effect on folding; nonnative isomers of other peptide groups may not affect folding at all.
3. Chemical reactions
Owing to its resonance stabilization, the peptide bond is relatively unreactive under physiological conditions, even less than similar compounds such as esters. Nevertheless, peptide bonds can undergo chemical reactions, usually through an attack of an electronegative atom on the carbonyl carbon, breaking the carbonyl double bond and forming a tetrahedral intermediate. This is the pathway followed in proteolysis and, more generally, in N-O acyl exchange reactions such as those of inteins. When the functional group attacking the peptide bond is a thiol, hydroxyl or amine, the resulting molecule may be called a cyclol or, more specifically, a thiacyclol, an oxacyclol or an azacyclol, respectively.
4. References
• Pauling L. (1960) The Nature of the Chemical Bond, 3rd. ed., Cornell University Press. ISBN 0-8014-0333-2
• Stein RL. (1993) “Mechanism of Enzymatic and Nonenzymatic Prolyl cis-trans Isomerization”, Adv. Protein Chem., 44, 1-24.
• Schmid FX, Layr LM, Mücke M and Schönbrunner ER. (1993) “Prolyl Isomerases: Role in Protein Folding”, Adv. Protein Chem., 44, 25-66.
• Fischer G. (1994) “Peptidyl-Prolyl cis/trans Isomerases and Their Effectors”, Angew. Chem. Int. Ed. Engl., 33, 1415-1436.
Peptide library
A peptide library is a newly developed technique for protein related study. A peptide library contains a great number of peptides that have a systematic combination of amino acids. Usually, peptide library is synthesized on solid phase, mostly on resin, which can be made as flat surface or beads. The peptide library provides a powerful tool for drug design, protein-protein interactions, and other biochemical as well as pharmaceutical applications.
Peptide sequence
Peptide sequence or amino acid sequence is the order in which amino acid residues, connected by peptide bonds, lie in the chain in Peptides and Proteins. The sequence is generally reported from the N-terminal end containing free amino group to the C-terminal end containing free carboxyl group. Peptide sequence is often called protein sequence if it represents the primary structure of a protein.
Sequence notation and applications
Many peptide sequences have been determined and recorded in sequence databases. These databases may use various notations to describe the peptide sequence. The full names of the amino acids are rarely given; instead, 3-letter or 1-letter abbreviations are usually recorded for conciseness.
Several deductions can be made from the sequence itself. Long stretches of hydrophobic residues may indicate transmembrane helices. These helices may indicate the peptide is a cell receptor. Certain residues indicate a beta sheet area. If full-length protein sequence is available, it is possible to estimate the isoelectric point of the protein. Methods for determining the peptide sequence include deduction from DNA sequence, Edman degradation, and mass spectrometry.
Techniques in sequence analysis can be applied to learn more about the peptide. These techniques generally consist of comparing the sequence to other sequences from sequence databases. Other sequences may have already been studied and determined to be significant. Findings about these sequences may be applicable to the sequence under investigation.
Peptide sequence tag
A peptide sequence tag is a piece of information about a peptide obtained by tandem mass spectrometry that can be used to identify this peptide in a protein database.[1][2][3]
1. Mass spectrometry
In general, peptides can be identified by fragmenting them in a mass spectrometer. For example, during collision-induced dissociation peptides collide with a gas within the mass spectrometer and break into pieces at their peptide bonds. The resulting fragment ions (called b-ions and y-ions) have mass differences corresponding to the residue masses of the respective amino acids. Thus, a tandem mass spectrum contains partial information about the amino acid sequence of the peptide. The peptide sequence tag approach, developed by Matthias Wilm and Matthias Mann at the EMBL,[4] uses this information to identify the peptide in a database. Briefly, a couple of masses is extracted from the spectrum in order to obtain the peptide sequence tag. This peptide sequence tag is a unique identifier of a specific peptide and can be used to find it in a database containing all possible peptide sequences.
2. Peptide fragment notation
Peptide fragmentation notation using the scheme of Roepstorff and Fohlman (1984).[5]A notation has been developed for indicating peptide fragments that arise from a tandem mass spectrum.[5] Peptide fragment ions are indicated by a, b, or c if the charge is retained on the N-terminus and by x, y or z if the charge is maintained on the C-terminus. The subscript indicates the number of amino acid residues in the fragment.
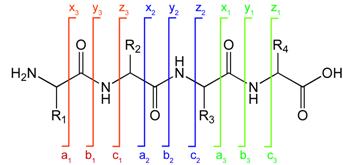
Peptide fragmentation notation using the scheme of Roepstorff and Fohlman (1984).[5]
3. References
1. Hardouin J (2007). “Protein sequence information by matrix-assisted laser desorption/ionization in-source decay mass spectrometry”. Mass spectrometry reviews 26 (5): 672-82.
2. Shadforth I, Crowther D, Bessant C (2005). “Protein and peptide identification algorithms using MS for use in high-throughput, automated pipelines”. Proteomics 5 (16): 4082-95.
3. Mørtz E, O’Connor PB, Roepstorff P, Kelleher NL, Wood TD, McLafferty FW, Mann M (1996). “Sequence tag identification of intact proteins by matching tanden mass spectral data against sequence data bases”. Proc. Natl. Acad. Sci. U.S.A. 93 (16): 8264-7.
4. Mann M, Wilm M (1994). “Error-tolerant identification of peptides in sequence databases by peptide sequence tags”. Anal. Chem. 66 (24): 4390-9.
5. Roepstorff P, Fohlman J (1984). “Proposal for a common nomenclature for sequence ions in mass spectra of peptides”. Biomed. Mass Spectrom. 11 (11): 601.
Peptide mass fingerprinting
Peptide mass fingerprinting (PMF) (also known as protein fingerprinting) is an analytical technique for protein identification that was developed 1993 by several groups independently (1-5). In short, the unknown protein of interest is cleaved into peptides by a protease such as Trypsin. The collection of peptides resulting from this cleavage comprise a unique identifier of the unknown protein. The absolute masses of the (still unknown) peptides are accurately measured with a mass spectrometer such as MALDI-TOF or ESI-TOF(6). These masses are then in silico compared to either a database containing known protein sequences or even the genome. Computer programs translate the known genome of the organism into proteins, then theoretically cut the proteins into peptides with the same protease (for example trypsin), and calculate the absolute masses of the peptides from each protein. They then compare the masses of the peptides of the unknown protein to the theoretical peptide masses of each protein encoded in the genome. The results are statistically analyzed to find the best match. The great advantage is that only the masses of the peptides have to be known (so de novo sequencing is not necessary). A disadvantage is that the protein sequence has to be present in the database of interest. Additionally most PMF algorithms assume the peptides come from a single protein(7). The presence of a mixture can significantly complicate the analysis and potentially compromise the results. Typical for the PMF based protein identification is the requirement for an isolated protein. Mixtures exceeding a number of 2-3 proteins typically require the additional use of MS/MS based protein identification to achieve sufficient specificity of identification (6). Therefore, the typical PMF samples are isolated proteins from Two-dimensional gel electrophoresis (2D gels) or isolated SDS-PAGE bands. Additional analyses by MS/MS can either be direct, e.g., MALDI-TOF/TOF analysis or downstream nanoLC-ESI-MS/MS analysis of gel spot eluates (7-9).
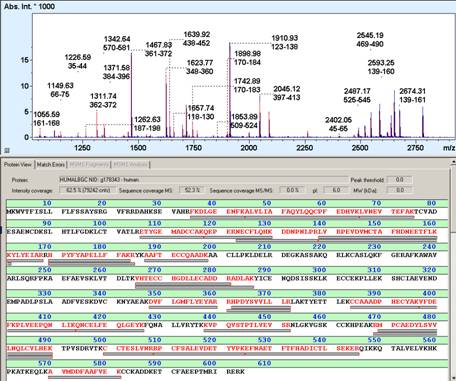
1. Sample preparation
Protein samples can be derived from SDS-PAGE (7) and are then subject to some chemical modifications. Disulfide bridges in proteins are reduced and cysteine amino acids are carboxymethylated chemically or acrylamidated during the gel electrophoresis.
Then the proteins are cut into several fragments using proteolytic enzymes such as trypsin, chymotrypsin or Glu-C. A typical sample:protease ratio is 50:1. The proteolysis is typically carried out overnight and the resulting peptides are extracted with acetonitrile and dried under vacuum. The peptides are then dissolved in a small amount of distilled water and are ready for mass spectrometric analysis.
2. Mass spectrometric analysis
The digested protein can be analyzed with different types of mass spectrometers such as ESI-TOF or MALDI-TOF. MALDI-TOF is often the preferred instrument because it allows a high sample throughput and several proteins can be analyzed in a single experiment – if complemented by MS/MS analysis.
A small fraction of the peptide (usually 1 microliter or less) is pipetted onto a MALDI target and a chemical called a matrix is added to the peptide mix. The matrix molecules are required for the desorption of the peptide molecules. Matrix and peptide molecules co-crystallize on the MALDI target and are ready to be analyzed.
The target is inserted into the vacuum chamber of the mass spectrometer and the analysis of peptide masses is initiated by a pulsed laser beam which transfers high amounts of energy into the matrix molecules. The energy transfer is sufficient to promote the transition of matrix molecules and peptides from the solid state into the gas state. Then the molecules become accelerated in the electric field of the mass spectrometer and fly towards an ion detector where their arrival is detected as an electric signal. Their mass is proportional to their time of flight (TOF) in the drift tube and can be calculated accordingly.
3. Computational analysis
The mass spectrometrical analysis produces a list of molecular weights which is often called peak list. The peptide masses are now compared to huge databases such as Swissprot, Genbank which contain protein sequence information. Software programs (see web resources, refs in 9) cut all these proteins into peptides with the same enzyme used in the chemical cleavage (for example trypsin). The absolute mass of all these peptides is then theoretically calculated. A comparison is made between the peak list of measured peptide masses and all the masses from the calculated peptides. The results are statistically analyzed and possible matches are returned in a results table.
4. References
1. Pappin DJ, Hojrup P, Bleasby AJ. 1993. Rapid identification of proteins by peptide-mass fingerprinting Curr Biol. Jun 1;3(6):327-32.
2. Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. 1993 Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases Proc Natl Acad Sci U S A. Jun 1;90(11):5011-5.
3. Mann M, Højrup P, Roepstorff P. 1993. Use of mass spectrometric molecular weight information to identify proteins in sequence databases Biol Mass Spectrom. Jun 22(6):338-45.
4. James P, Quadroni M, Carafoli E, Gonnet G. 1993.Protein identification by mass profile fingerprinting Biochem Biophys Res Commun. 1993 Aug 31;195(1):58-64.
5. Yates JR 3rd, Speicher S, Griffin PR, Hunkapiller T. 1993. Peptide mass maps: a highly informative approach to protein identification Anal Biochem. Nov. 1;214(2):397-408.
6. Clauser KR, Baker PR, Burlingame AL. 1999 Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 71(14):2871-82.
7. Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M. 1996 Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels Proc Natl Acad Sci U S A. 93(25):14440-5.
8. Wang W, Sun J, Nimtz M, Deckwer WD, Zeng 2003 Protein identification from two-dimensional gel electrophoresis analysis of Klebsiella pneumoniae by combined use of mass spectrometry data and raw genome sequences Proteome Sci. 1(1):6.
9. Hufnagel P, Rabus R.2006 Mass spectrometric identification of proteins in complex post-genomic projects. Soluble proteins of the metabolically versatile, denitrifying ‘Aromatoleum’ sp. strain EbN1. J Mol Microbiol Biotechnol. 11(1-2):53-81.
5. Web resources
6. External links
Antimicrobial peptides
Antimicrobial peptides (also called host defence peptides) are an evolutionarily conserved component of the innate immune response and are found among all classes of life.
These peptides are potent, broad spectrum antibiotics which demonstrate potential as novel therapeutic agents. Antimicrobial peptides have been demonstrated to kill Gram negative and Gram positive bacteria (including strains that are resistant to conventional antibiotics), mycobacteria (including Mycobacterium tuberculosis), enveloped viruses, fungi and even transformed or cancerous cells. Unlike the majority of conventional antibiotics it appears as though antimicrobial peptides may also have the ability to enhance immunity by functioning as immunomodulators.
1. Structure
Antimicrobial peptides are short proteins, generally between 12 and 50 amino acids long (although larger proteins with similar properties such as lysozyme are often classified as antimicrobial peptides). These peptides include two or more positively charged residues provided by arginine, lysine or, in acidic environments, histidine, and a large proportion (generally >50%) of hydrophobic residues.[1][2] The secondary structures of these molecules follow 4 themes, including i) α-helical, ii) β-stranded due to the presence of 2 or more disulphide bonds, iii) β-hairpin or loop due to the presence of a single disulphide bond and/or cyclization of the peptide chain, and iv) extended. Many of these peptides are unstructured in free solution, and fold into their final configuration upon partitioning into biological membranes. The ability to associate with membranes is a definitive feature of antimicrobial peptides [3] although membrane permeabilisation is not necessary. These peptides have a variety of antimicrobial activities ranging from membrane permeabilization to action on a range of cytoplasmic targets.
2. Antimicrobial Activities
The modes of action by which antimicrobial peptides kill bacteria is varied and includes disrupting membranes, interfering with metabolism, and targeting cytoplasmic components. In many cases the exact mechanism of killing is not known. In contrast to many conventional antibiotics these peptides appear to be bacteriocidal (bacteria killer) instead of bacteriostatic (bacteria growth inhibitor). In general the antimicrobial activity of these peptides is determined by measuring the minimal inhibitory concentration (MIC), which is the lowest concentration of drug that reduces growth by more than 50%.[4]
3. Immunomodulatory Activities
In addition to killing bacteria directly they have been demonstrated to have a number of immunomodulatory functions that may be involved in the clearance of infection, including the ability to alter host gene expression, act as chemokines and/or induce chemokine production, inhibiting lipopolysaccharide induced pro-inflammatory cytokine production, promoting wound healing, and modulating the responses of dendritic cells and cells of the adaptive immune response. Animal models indicate that host defence peptides are crucial for both prevention and clearance of infection. It appears as though many peptides initially isolated as and termed “antimicrobial peptides” have been shown to have more significant alternative functions in vivo.
4. Therapeutic Potential
These peptides are excellent candidates for development as novel therapeutic agents and complements to conventional antibiotic therapy because in contrast to conventional antibiotics they do not appear to induce antibiotic resistance while they generally have a broad range of activity, are bacteriocidal as opposed to bacteriostatic and require a short contact time to induce killing. A number of naturally occurring peptides and their derivatives have been developed as novel anti-infective therapies for conditions as diverse as oral mucositis, lung infections associated with cystic fibrosis (CF) and topical skin infections.
5. Notes and references
1. Papagianni, M. 2003. Ribosomally synthesized peptides with antimicrobial properties: biosynthesis, structure, function, and applications. Biotechnol Adv 21:465.
2. Sitaram, N., and R. Nagaraj. 2002. Host-defense antimicrobial peptides: importance of structure for activity. Curr Pharm Des 8:727.
3. Hancock, R. E. W., and A. Rozek. 2002. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol Lett 206:143.
4. National Committee of Laboratory Safety and Standards (NCLSS) as published in Amsterdam, D. 1996. Susceptibility testing of Antimicrobials in liquid media. In “Antibiotics in Laboratory Medicine”, Lorian, V., ed. Fourth Edition, pp.52-111. Williams and Wilkins, Baltimore
6. External links
• A good basic overview of cationic peptides at scq.ubc.ca
• Review with helpful links at ejbiotechnology.info
• Antimicrobial Peptide Database
Beta-peptide
β-peptides consist of β amino acids, which have their amino group bonded to the β carbon rather than the α carbon as in the 20 standard biological amino acids. The only commonly naturally occurring β amino acid is β-alanine; although it is used as a component of larger bioactive molecules, β-peptides in general do not appear in nature. For this reason β-peptide-based antibiotics are being explored as ways of evading antibiotic resistance.
1. Chemical structure and synthesis
In α amino acids (molecule in Figure 1), both the carboxylic acid group (red) and the amino group (blue) are bonded to the same carbon, termed the α carbon (C-α) because it is one atom away from the carboxylate group. In β amino acids, the amino group is bonded to the β carbon (C-β), which is found in most of the 20 standard amino acids. Only glycine lacks a β carbon, which means that there is no β-glycine molecule.
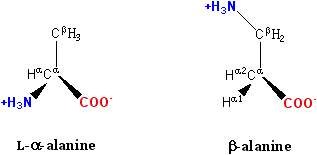
Figure 1: β-alanine, an example of a β amino acid. The amino group attaches not to the α carbon but to the β carbon, which in this case is the sidechain methylene.
The chemical synthesis of β amino acids can be challenging, especially given the diversity of functional groups bonded to the β carbon and the necessity of maintaining chirality. In the alanine molecule shown, the β carbon is achiral; however, most larger amino acids have a chiral Cβ atom. A number of synthesis mechanisms have been introduced to efficiently form β amino acids and their derivatives[1][2].
2. Secondary Structure
Because the backbones of β-peptides are longer than those of peptides that consist of α-amino acids, β-peptides form different secondary structures. The alkyl substituents at both the α and β positions in a β amino acid favor a gauche conformation about the bond between the α-carbon and β-carbon. This also affects the thermodynamic stability of the structure.
Many types of helix structures consisting of β-peptides have been reported. These conformation types are distinguished by the number of atoms in the hydrogen-bonded ring that is formed in solution; 8-helix, 10-helix, 12-helix, 14-helix, and 10/12-helix have been reported. Generally speaking, β-peptides form a more stable helix than α-peptides [3].
3. Clinical potential
β-peptides are stable against proteolytic degradation in vitro and in vivo, an important advantage over natural peptides in the preparation of peptide-based drugs [4]. β-peptides have been used to mimic natural peptide-based antibiotics such as magainins, which are extremely powerful but difficult to use as drugs because they are degraded by proteolytic enzymes in the body [5].
4. References
1. Gademann K, Hintermann T, Schreiber JV. (1999). “Beta-peptides: twisting and turning.”, Curr Med Chem Oct;6(10):905-25. [6].
2. Basler B, Schuster O, Bach T. (2005). Conformationally constrained beta-amino acid derivatives by intramolecular [2 + 2]-photocycloaddition of a tetronic acid amide and subsequent lactone ring opening. J Org Chem 70(24):9798-808. [7].
3. Murray JK, Farooqi B, Sadowsky JD, Scalf M, Freund WA, Smith LM, Chen J, Gellman SH. (2005). Efficient synthesis of a beta-peptide combinatorial library with microwave irradiation. J Am Chem Soc127(38):13271-80. [8]
4. Beke T, Somlai C, Perczel A. (2006). “Toward a rational design of beta-peptide structures.”, J Comp Chem Jan 15;27(1):20-38. [9].
5. Porter EA, Weisblum B, Gellman SH. (2002). Mimicry of host-defense peptides by unnatural oligomers: antimicrobial beta-peptides. J Am Chem Soc 124(25):7324-30.
Cyclotides
Cyclotides are small disulfide rich peptides isolated from plants (1). Typically containing 28-37 amino acids, they are characterized by their head-to-tail cyclised peptide backbone and the interlocking arrangement of their three disulfide bonds. These combined features have been termed the cyclic cystine knot (CCK) motif (Figure 1). To date, over 100 cyclotides have been isolated and characterized from species of the Rubiaceae, Violaceae and Cucurbitaceae plant families.
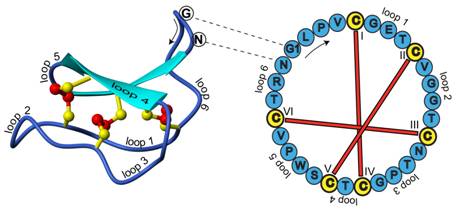
Figure 1. Structure and sequence of the prototypic cyclotide kalata B1
Contents
1. Cyclotide structure
2. Biological significance
3. A Serendipitous Discovery
4. Cyclotide amino-acid sequences
5. Biosynthesis of cyclotides
6. Applications
7. References
1. Cyclotide structure
Cyclotides have a well-defined three-dimensional structure as a result of their interlocking disulfide bonds and cyclic peptide backbone. Backbone loops and selected residues are labeled on the structure to help orientation. The amino acid sequence (single letter amino acid representation) for this peptide is indicated on the sequence diagram to the right. One of the interesting features of cyclic peptides is that knowledge of the peptide sequence does not reveal the ancestral head and tail; knowledge of the gene sequence is required for this (2). In the case of kalata B1 the indicated glycine (G) and asparagine (N) amino acids are the terminal residues that are linked in a peptide bond to cyclise the peptide.
2. Biological significance
Cyclotides have been reported to have a wide range of biological activities, including anti-HIV, insecticidal, anti-tumour, antifouling, anti-microbial, hemolytic, neurotensin antagonism, trypsin inhibition, and uterotonic activities (3-5). An ability to induce uterine contractions was what prompted the initial discovery of kalata B1 (6).
The potent insecticidal activity of cyclotides kalata B1 and kalata B2 has prompted the belief that cyclotides act as plant host-defence agents. The observations that dozens or more cyclotides may be present in a single plant and the cyclotide architecture comprises a conserved core onto which a series of hypervariable loops is displayed suggest that, cyclotides may be able to target many pests/pathogens simultaneously.
3. A Serendipitous Discovery
During a Red Cross relief mission in the Congo during the 1960s, a Norwegian doctor, Lorents Gran, noted that during labor African women used a medicinal tea made from the leaves of the plant Oldenlandia affinis to induce labor and facilitate childbirth (8). The active ingredient was later determined to be a peptide, named kalata B1, after the traditional name for the native medicine, kalata-kalata. Although in vivo studies in rats confirmed the uterotonic activity of the purified peptide, it was another 20 years before the cyclic cystine knot motif and structure of the purified peptide were elucidated (9).
4. Cyclotide amino-acid sequences
Analysis of the suite of known cyclotides reveals many sequence homologies that are important for understanding their unique physico-chemical properties and bioactivities. Table 1 presents a selection of cyclotides.
The cyclotides fall into two main structural subfamilies. Moebius cyclotides, the less common of the two, contain a cis-proline in loop 5 that induces a local 180º backbone twist (hence likening it to a Möbius strip, whereas bracelet cyclotides do not. There is smaller variation in sequences within these subfamilies than between them. A third subfamily of cyclotides are trypsin inhibitors and are more homologous to a family of non-cyclic trypsin inhibitors from squash plants known as knottins (10) than they are to the other cyclotides.
It is convenient to discuss sequences in terms of the backbone segments, or loops, between successive cysteine residues. The six cysteine residues are absolutely conserved throughout the cyclotide suite and presumably contribute to the preservation of the CCK motif. Although the cysteines appear essential to maintaining the overall fold, several other residues that are highly conserved in cyclotides are thought to provide additional stability (11).
Throughout the known cyclotides loop 1 is the most conserved. Apart from the six cysteine residues, the glutamic acid and serine/threonine residues of loop 1 are the only residues to have 100% identity across the bracelet and Möbius subfamilies. Furthermore the remaining residue of this loop exhibits only a conservative change i.e. glycine/alanine. This loop is believed to play an important role in stabilizing the cyclotide structure through hydrogen bonding with residues from loops 3 and 5.
Loops 2-6 also have highly conserved features, including the ubiquitous presence of just a single amino acid in loop 4 that is likely involved in sidechain-sidechain hydrogen bonding. Other conserved residues include a hydroxyl-containing residue in loop 3, a glycine residue in the final position of loop 3, a basic and a proline residue in the penultimate position in loop 5 of bracelet and Möbius cyclotides respectively, and an asparagine (or occasionally aspartic acid) residue at the putative cyclisation (2,7,12) point in loop 6. It is of interest to note that not only are certain residues highly conserved, but the backbone and side chain angles are as well.
With recent screening programs suggesting that the number of cyclotide sequences may soon reach the thousands (13), a database, CyBase, has been developed that offers the opportunity for comparisons of sequences and activity data for cyclotides. Several other families of circular proteins are known in bacteria, plants and animals and are also included in CyBase (14).
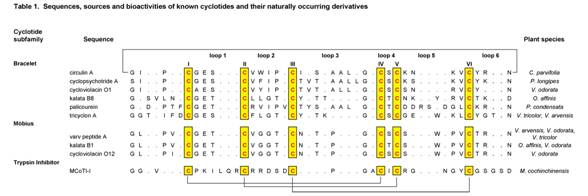
Table 1. A selection of cyclotide sequences and sources
5. Biosynthesis of cyclotides
Plants are a rich source of cyclic peptides, with the vast majority of these molecules being produced via non-ribosomal biosynthetic pathways. In contrast, the cyclotides are gene-coded products generated via processing of a larger precursor protein (2). The gene for the first such precursor is Oak1 (Oldenlandia affinis kalata clone number 1), which was shown to be responsible for the synthesis of kalata B1 (7). Figure 2 illustrates the generic configuration of the precursor protein, which consist of an endoplasmic reticulum signal sequence, a non-conserved pro-region, a highly conserved region known as the N-terminal repeat (NTR), the mature cyclotide domain and finally a short hydrophobic C-terminal tail. The cyclotide domain may contain either one cyclotide sequence, as in the case of Oak1, or multiple copies separated by additional NTR sequences as seen for Oak2 and Oak4. In precursor proteins containing multiple cyclotide domains these can either be all identical sequences, as is the case for Oak4, or they can be different cyclotides as in Oak2 which contains sequences corresponding to kalata B3 and B6.
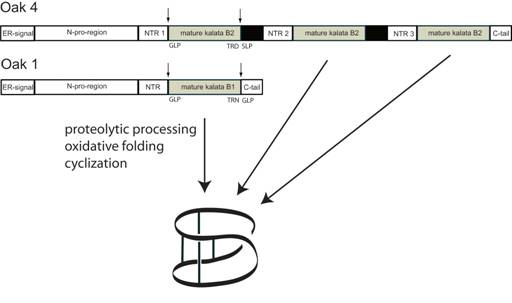
Figure 2. Schematic representation of the cyclotide precursors Oak1 and Oak4 and proposed mechanism of biosynthesis (15).
6. Applications
The remarkable stability of cyclotides means that they have an exciting range of potential applications centred on either their intrinsic biological activities or the possibility of using the CCK motif as a scaffold for stabilizing biologically active epitopes (16). Interest in these has recently intensified with the publications of a chemical methodology capable of synthetically producing cyclotides with high yields (17), and the amenability of the CCK framework to amino-acid substitutions (18). But for molecules to be useful in a therapeutic setting they require useful biopharmaceutical characteristics such as resistance to proteolysis and membrane permeability. A recent study on related cystine knot proteins as drug candidates showed that cystine knots do permeate well through rat small intestinal mucosa relative to non-cystine knot peptide drugs such as insulin and bacitracin (19). Furthermore, enzymatic digestion of cystine knot peptide drugs was associated with only a few proteases and it was suggested that this limitation may be overcome by mutating out particular cleavage sites. Thus, certain cystine knot proteins satisfy the basic criteria for drug delivery and represent exciting novel candidates as scaffolds for peptide drug delivery (19). The diverse range of intrinsic activities of cyclotides also continues to hold promise for a wide range of applications in the agricultural fields.
7. References
1. Craik, D. J., Daly, N. L., Bond, T., and Waine, C. (1999). J. Mol. Biol. 294, 1327-1336
2. Dutton, J. L., Renda, R. F., Waine, C., Clark, R. J., Daly, N. L., Jennings, C. V., Anderson, M. A., and Craik, D. J. (2004). J. Biol. Chem. 279, 46858-46867
3. Craik, D. J., Daly, N. L., Mulvenna, J., Plan, M. R., and Trabi, M. (2004). Curr. Prot. Pept. Sci. 5, 297-315
4. Göransson, U., Sjogren, M., Svangard, E., Claeson, P., and Bohlin, L. (2004). J. Nat. Prod. 67, 1287-1290
5. Gustafson, K. R., McKee, T. C., and Bokesch, H. R. (2004). Curr. Protein Pept. Sci. 5, 331-340
6. Gran, L. (1970). Medd. Nor. Farm. Selsk. 12, 173-180
7. Jennings, C., West, J., Waine, C., Craik, D., and Anderson, M. (2001). Proc. Natl. Acad. Sci. U S A 98, 10614-10619
8. Gran, L., Sandberg, F., and Sletten, K. (2000). J. Ethnopharmacol. 70, 197-203
9. Saether, O., Craik, D. J., Campbell, I. D., Sletten, K., Juul, J., and Norman, D. G. (1995). Biochemistry 34, 4147-4158
10. Chiche, L., Heitz, A., Gelly, J. C., Gracy, J., Chau, P. T., Ha, P. T., Hernandez, J. F., and Le-Nguyen, D. (2004). Curr. Protein Pept. Sci. 5, 341-349
11. Rosengren, K. J., Daly, N. L., Plan, M. R., Waine, C., and Craik, D. J. (2003). J. Biol. Chem. 278, 8606-8616
12. Ireland, D. C., Colgrave, M. L., Nguyencong, P., Daly, N. L., and Craik, D. J. (2006). J. Mol. Biol. 357, 1522-1535
13. Simonsen, S. M., Sando, L., Ireland, D. C., Colgrave, M. L., Bharathi, R., Göransson, U., and Craik, D. J. (2005). Plant Cell 17, 3176-3189
14. Craik, D. J. (2006). Science 311, 1563-1564
15. Gruber, C. W., Čemažar, M., Anderson, M. A., and Craik, D. J. (2006). Toxicon 49, 561-575
16. Craik, D. J., Simonsen, S., and Daly, N. L. (2002). Curr. Opin. Drug Discov. Devel. 5, 251-260
17. Gunasekera, S., Daly, N. L., Anderson, M. A., and Craik, D. J. (2006). IUBMB Life 58, 515-524
18. Craik, D. J., Čemažar, M., and Daly, N. L. (2006). Curr. Opin. Drug Discov. Devel. 9, 251-260
19. Werle, M., Schmitz, T., Huang, H. L., Wentzel, A., Kolmar, H., and Bernkop-Schnurch, A. (2006). J. Drug Target 14, 137-146
20. Shenkarev ZO, Nadezhdin KD, Sobol VA, Sobol AG, Skjeldal L, ArsenievAS. 2006. Conformation and mode of membrane interaction in cyclotides. FEBS J. 273: 2658-2672.
Glycopeptide antibiotic
Glycopeptide antibiotics are a class of antibiotic drugs. They consist of a glycosylated cyclic or polycyclic nonribosomal peptide. Important glycopeptide antibiotics include vancomycin, teicoplanin, ramoplanin, and decaplanin.
This class of drugs inhibit the synthesis of cell walls in susceptible microbes by inhibiting peptidoglycan synthesis. They bind to the amino acids within the cell wall preventing the addition of new units to the peptidoglycan. In particular they bind to acyl-D-alanyl-D-alanine in peptidoglycan. The D stands for the dextro stereoisomer of the amino acid, which is signigficant as amino acids are normal L or levo stereoisomers. Due to their toxicity, their use is restricted to those patients who are critically ill or who have a demonstrated hypersensitivity to the β-lactams. Principally effective against gram positive cocci, they exhibit a narrow spectrum of action. They are the last effective line of defense for cases of Methicillin-resistant Staphylococcus aureus, however vancomycin-resistant MRSA has been seen in some countries, which could present a problem to treatment. They are bactericidal against most species, but are only bacteriostatic against the enterococci.
Teicoplanin was discovered in the early 1990s. It is more lipophillic than vancomycin, as it has more fatty acid chains. It is considered to be 50 to 100 times more lipophillic than vancomycin. Teicoplanin has an increased half life compared to vancomycin, as well as having better tissue penetration. It can be two to four times more active than vancomycin, but it does depend upon the organism. Teicoplanin is more acidic, forming water soluble salts, so it can be give intramuscularly. Teicoplanin is much better at penetrating into leucocytes and phagocytes than vancomycin.
Some tissues are not penetrated very well by glycopeptides, and they don’t penetrate into the CSF.
Vancomycin is usually given intravenously, as an infusion, and can cause tissue necrosis and phlebitis at the injection site if given too rapidly. Indeed pain at site of injection is a common adverse event. One of the side effects is ‘Red man syndrome’, an idiosynchratic reaction to bolus, caused by histamine release. Some other side effects of vancomycin are nephrotoxicity including renal failure and interstitial nephritis, blood disorders including neutropenia and deafness, which is reversible once therapy has stopped. Oral preparations are available, however thay aren’t absorbed from the lumen of the gut, so are of no use in treating systemic infections. The oral preparations are formulated for the treatment of infections within the Gastro-Intestinal tract, Clostridium difficile for example.
Over 90% of the dose is excreted in the urine, therefore there is a risk of accumulation in patients with renal impairment, so therapeutic drug monitoring (TDM) is recommended.
Nonribosomal peptide
Nonribosomal peptides (NRP) are a class of secondary metabolites, usually produced by microorganisms like bacteria and fungi. These are also often found in higher organisms, such as nudibranchs but they are thought to be made by bacteria inside these organisms. Note that although there are many peptides which are not made on the ribosome, nonribosomal peptide typically refers to a very specific set of these as discussed in this article.
Unlike polypeptides synthesized on the ribosome, these peptides are synthesized by nonribosomal peptide synthetases (NRPS) from amino acids. NRPS can be thought of as preassembled, modular, molecular factories. Unlike the ribosome, which is fed an mRNA code and can make an arbitrary sequence of peptides, an NRPS does not accept a code and is preset to make one peptide. As a class, NRPS can make a wider diversity of peptides than can ribosomes.
NRPs often have a cyclic and/or branched structure, contain non-proteinogenic amino acids including D-amino acids, carry modifications like N-methyl and N-formyl groups, or are glycosylated, acylated, halogenated, or hydroxylated. Cyclization of amino acids against the peptide “backbone” is often performed, resulting in oxazolines and thiazolines; these can be further oxidized or reduced. Occasionally dehydration is performed on serines resulting on dehydroalanine. This is just a sampling of the various manipulations and variations that NRPS can perform. NRPs are often dimers or trimers of identical sequences chained together or cyclized, or even branched.
Nonribosomal peptides are structurally a very diverse family of natural products with an extremely broad range of biological activities and pharmacological properties. They are often toxins, siderophores, or pigments. Nonribosomal peptide antibiotics, cytostatics, and immunosuppressants are in commercial use.
1. Examples
• Antibiotics
• Bacitracin
• Vancomycin
• Tyrocidin
• Gramicidine
• Thiostrepton
• Antibiotics precursors
• ACV-Tripeptide
• Cytostatics
• Epothilone
• Bleomycin
• Immunosuppressants
• Ciclosporin (Cyclosporine A)
• Siderophores
• Enterobactin
• Myxochelin A
• Pigments
• Indigodin
• Toxins
• Microcystins and
• Nodularins, cyanotoxins from cyanobacteria.
• α-amanitine from Amanita phalloides (Death Cap)
• Nitrogen storage polymers
• Cyanophycin – produced by some cyanobacteria
2. Biosynthesis
Nonribosomal peptides are synthesized by one or more specialized nonribosomal peptide-synthetase (NRPS) enzymes. The NRPS genes for a certain peptide are usually organized in one operon in bacteria and in gene clusters in eukaryotes. The enzymes are organized in modules that are responsible for the indroduction of one additional amino acid. Each module consists of several domains with defined functions, separated by short spacer regions of about 15 amino acids.
The biosynthesis of nonribosomal peptides shares similarities with the polyketide and fatty acid biosynthesis. Due to these structural and mechanistic similarities some nonribosomal peptide synthetases contain Polyketide synthase modules for the insertion of acetate or propionate derived subunits into the peptide chain.
2.1. Modules
The order of modules and domains of a complete nonribosomal peptide synthetase is as follows:
• Initiation or Starting module: [F/NMe]-A-PCP-
• Elongation or Extending modules: -(C/Cy)-[NMe]-A-PCP-[E]-
• Termination or Releasing module: -(TE/R)
(Order: N-terminus to C-terminus; []: optionally; (): alternatively)
2.2. Domains
• F: Formylation (optional)
• A: Adenylation (required in a module)
• PCP: Thiolation and Peptide Carrier Protein with attached 4′-phospho-pantethein (required in a module)
• C: Condensation forming the amide bond (required in a module)
• Cy: Cylization into thiazoline or oxazolines (optional)
• Ox: Oxidation of thiazolines or oxazolines to thiazoles or oxazoles (optional)
• Red: Reduction of thiazolines or oxazolines to thiazolidines or oxazolidines (optional)
• E: Epimerization into D-amino acids (optional)
• TE: Termination by a thio-esterase (only found once in a NRPS)
• R: Reduction to terminal aldehyde or alcohol (optional)
2.3. Starting stage
• Loading: The first amino acid is activated with ATP as a mixed acyl-phosphoric acid anhydride with AMP by the A-domain and loaded onto the serine-attached 4′-phospho-pantethein (4’PP) sidechain of the PCP-domain catalyzed by the PCP-domain (thiolation) .
• Sometimes the amino group of the bound amino acid is formylated by an F-domain or methylated by an NMe-domain.
2.4. Elongation stages
• Loading: Analogous to the starting stage, each module loads its specific amino acid onto its PCP-domain.
• Condensation: The C-domain catalyzes the amide bond formation between the thioester group of the growing peptide chain from the previous module with the amino group of the current module. The extended peptide is now attached to the current PCP-domain.
• Condensation-Cyclization: Sometimes the C-domain is replaced by a Cy-domain which, in addition to the amide bond formation, catalyzes the reaction of the serine, threonine, or cysteine sidechain with the amide-N, thereby forming oxazolidines and thiazolidine, respectively.
• Epimerization: Sometimes an E-domain epimerizes the innermost amino acid of the peptide chain into the D-configuration.
• This cycle is repeated for each elongation module.
2.5. Termination stage
• Termination: The TE-domain (thio-esterase domain) hydrolyzes the completed polypeptide chain from the ACP-domain of the previous module, thereby often forming cyclic amides (lactams) or cyclic esters (lactones).
• Alternatively, the peptide can be released by an R-domain that reduces the thioester bond to terminal aldehyde or alcohol.
2.6. Processing
The final peptide is often modified, e.g. by glycosylation, acylation, halogenation, or hydroxylation. The responsible enzymes are usually associated to the synthetase complex and their genes are organized in the same operons or gene clusters.
2.7. Priming and Deblocking
To become functional, the 4′-phospho-pantethein sidechain of acyl-CoA molecules has to be attached to the PCP-domain by 4’PP transferases (Priming) and the S-attached acyl group has to be removed by specialized associated thioesterases (TE-II) (Deblocking).
2.8. Substrate specificities
Most domains have a very broad substrate specificity and usually only the A-domain determines which amino acid is incorporated in a module. Ten amino acids have been identified that control substrate specificity and can be considered the ‘codons’ of nonribosomal peptide synthesis. The condensation C-domain is also believed to have substrate specificity, especially if located behind an epimerase E-domain containing module where it functions as a ‘filter’ for the epimerized isomer.
3. Mixed with Polyketides
Due to the similarity with polyketide synthetases (PKS), many secondary metabolites are in fact fusions of NRPs and polyketides. This essentially occurs when PK modules follow NRP modules, and vice versa. There is high degree of similarity between the PCP domains of both types of sythetases, although the mechanism of condensation is different from a chemical standpoint (claisen vs. transamidation).
Epothilone
4. Literature
• “Nonribosomal peptides: from genes to products” by Dirk Schwarzer, Robert Finking, and Mohamed A. Marahiel in Nat. Prod. Rep. 20(3):275-287 (2003)
• “Modular Peptide Synthetases Involved in Nonribosomal Peptide Synthesis” by Mohamed A. Marahiel, Torsten Stachelhaus, and Henning D. Mootz in Chem. Rev. 97(7):2651-2673 (1997)
Signal peptide
A signal peptide is a short (3-60 amino acids long) peptide chain that directs the post-translational transport of a protein. Signal peptides may also be called targeting signals, signal sequences, transit peptides, or localization signals.
The amino acid sequences of signal peptides direct proteins (which are synthesized in the cytosol) to certain organelles such as the nucleus, mitochondrial matrix, endoplasmic reticulum, chloroplast, apoplast and peroxisome. Some signal peptides are cleaved from the protein by signal peptidase after the proteins are transported.
1. ER signal peptide
An endoplasmic reticulum signal peptide is the best characterised signal peptide. It exists at the amino terminal of a protein. The protein is guided to the ER by a signal-recognition particle, which moves between the ER and the cytoplasm. It binds to the signal peptide. The SRP binds to the signal peptide as soon as it is synthesised and extruded from the ribosome. This causes a pause in protein synthesis, most probably allowing the ribosome-SRP complex time to bind to the SRP receptor on the target ER membrane. The SRP protein is thought to be a regulatory GTP protein. Conformational changes may therefore lead to the SRP release. The protein may then be threaded through the ER membrane by a translocator pore.
2. Nuclear signal peptides
A nuclear localization signal (NLS) is a signal peptide directing to the nucleus and is often a unit consisting of plus-charged amino acids. The NLS normally is located inside the peptide chain. Almost all proteins that are transported to the endoplasmic reticulum have a sequence consisting of 5-10 hydrophobic amino acids on the N-terminus. Most of these proteins are transported from the endoplasmic reticulum to the Golgi apparatus. If these proteins have a particular 4-amino-acids sequence on the C-terminus, these proteins stay in the endoplasmic reticulum.
The nucleolus within the nucleus can be targeted with a sequence called a nucleolar localization signal (abbreviated NoLS or NOS).
The signal peptide that directs to the mitochondrial matrix has a sequence consisting of an alternating pattern with a few hydrophobic amino acids and a few plus-charged amino acids form. It is usually called the mitochondrial targeting signal (MTS).
There are two types of signal peptides directing to peroxisome, which are called peroxisomal targeting signals (PTS). One is PTS1, which is made of three amino acids on the C-terminus. The other is PTS2, which is made of a 9-amino-acid sequence often present on the N-terminus of the protein.
3. Types
Following is a list of types of signal peptides:
• N-terminus signal peptides often target the mitochondrial matrix, endoplasmic reticulum and peroxisome.
• C-terminus signal peptides often target the peroxisome.
4. External links
Microcystin
Microcystins are cyclic nonribosomal peptides produced by cyanobacteria. They are cyanotoxins and can be very toxic for plants and animals including humans. Their hepatotoxicity may cause serious damage to the liver.
Microcystins consist of several uncommon non-proteinogenic amino acids such as dehydroalanine derivatives and the special β-amino acid ADDA ((all-S,all-E)-3-Amino-9-methoxy-2,6,8-trimethyl-10-phenyldeca-4,6-diene acid).
There appears to be inadequate information to assess carcinogenic potential of microcystins by applying EPA Guidelines for Carcinogen Risk Assessment. A few studies suggest that there may be a relationship between liver and colorectral cancers and the occurrence of cyanobacteria in drinking water in China (Yu et al., 1989; Zhou et al., 2002). Evidence is, however, limited due to limited ability to accurately assess and measure exposure.
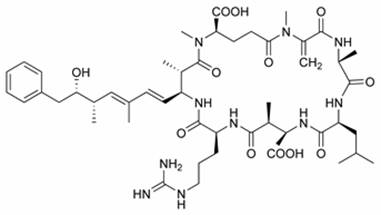
Chemical structure of Microcystin LR
Microcystin-LR is one of over 80 known toxic variants and is the most studied by biologists and ecologists. Microcystin containing ‘blooms’ are a problem worldwide, including China, Australia, the United States and much of Europe. Once ingested, microcystin travels to the liver, via the bile acid transport system, where most is stored; though some remains in the blood stream and may contaminate tissue. Microcystin binds covalently to protein phosphatases thus disrupting cellular control processes.
References
• NationalCenter for Environmental Assessment. Toxicological Reviews of Cyanobacterial Toxins: Microcystins LR, RR, YR and LA (NCEA-C-1765)
• Yu, S.-Z. 1989. Drinking water and primary liver cancer. In: Primary Liver Cancer, Z.Y. Tang, M.C. Wu and S.S. Xia, Ed. China Academic Publishers, New York, NY. p. 30-37 (as cited in Ueno et al., 1996 and Health Canada, 2002).
• Zhou, L., H. Yu and K. Chen. 2002. Relationship between microcystin in drinking water and colorectal cancer. Biomed. Environ. Sci. 15(2):166-171.
Peptide nucleic acid
Peptide nucleic acid (PNA) is a chemical similar to DNA or RNA. PNA is not known to occur naturally in existing life on Earth but is artificially synthesized and used in some biological research and medical treatments.
DNA and RNA have a deoxyribose and ribose sugar backbone, respectively, whereas PNA’s backbone is composed of repeating N-(2-aminoethyl)-glycine units linked by peptide bonds. The various purine and pyrimidine bases are linked to the backbone by methylene carbonyl bonds. PNAs are depicted like peptides, with the N-terminus at the first (left) position and the C-terminus at the right.
Since the backbone of PNA contains no charged phosphate groups, the binding between PNA/DNA strands is stronger than between DNA/DNA strands due to the lack of electrostatic repulsion. Early experiments with homopyrimidine strands (strands consisting of only one repeated pyrimidine base) have shown that the Tm (“melting” temperature) of a 6-base thymine PNA/adenine DNA double helix was 31°C in comparison to an equivalent 6-base DNA/DNA duplex that denatures at a temperature less than 10°C. Mixed base PNA molecules are true mimics of DNA molecules in terms of base-pair recognition. PNA/PNA binding is stronger than PNA/DNA binding.
Synthetic peptide nucleic acid oligomers have been used in recent years in molecular biology procedures, diagnostic assays and antisense therapies. Due to their higher binding strength it is not necessary to design long PNA oligomers for use in these roles, which usually require oligonucleotide probes of 20-25 bases. The main concern of the length of the PNA-oligomers is to guarantee the specificity. PNA oligomers also show greater specificity in binding to complementary DNAs, with a PNA/DNA base mismatch being more destabilizing than a similar mismatch in a DNA/DNA duplex. This binding strength and specificity also applies to PNA/RNA duplexes. PNAs are not easily recognized by either nucleases or proteases, making them resistant to enzyme degradation. PNAs are also stable over a wide pH range.
It has been hypothesized that the earliest life on Earth may have used PNA as a genetic material due to its extreme robustness, simpler formation and possible spontaneous polymerization at 100°C (while water at standard pressure boils at this temperature, water at high pressure – as in deep ocean – boil at higher temperatures). If this is so, life transitioned to a DNA/RNA-based system only at a later stage[1] [2]. Evidence for this hypothesis is however far from conclusive. See RNA world hypothesis for related information.
References
1. Nelson, K.E., Levy, M., and Miller, S.L. Peptide nucleic acids rather than RNA may have been the first genetic molecule (2000) Proc. Natl. Acad. Sci. USA 97, 3868–3871.
2. Alberts, Johnson, Lewis, Raff, Roberts and Walter, Molecular Biology of the Cell, 4th Edition, Routledge, March, 2002, ISBN 0-8153-3218-1.
External links
• Peptide nucleic acids (PNAs), a chemical overview
• Recognition of chromosomal DNA by PNAs
• Alternative Nucleic Acid Analogues for Programmable Assembly: Hybridization of LNA to PNA
• The peptide nucleic acids (PNAs): a new generation of probes for genetic and cytogenetic analyses
