Peptide Modifications
This entry is from Wikipedia, the leading user-contributed encyclopedia.
• Fluorescein
• Fluorescent labeling
• Acetylation
• Amidation
• Palmitoylation
• Myristoylation
• Phosphorylation
• Glycosylation
• Biotinylation
• Farnesylation
Fluorescein
Fluorescein is a fluorophore commonly used in microscopy, in a type of dye laser as the gain medium, in forensics and serology to detect latent blood stains, and in dye tracing. Fluorescein has an absorption maximum at 494 nm and emission maximum of 521 nm (in water). Also, fluorescein has an isoabsorptive point (equal absorption for all pH values) at 460 nm. Fluorescein is also known as a color additive (D&C Yellow no. 7). The disodium salt form of fluorescein is known as D&C Yellow no. 8.
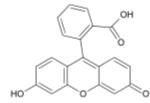
1. Chemical and physical properties
The fluorescence of this molecule is very high, and excitation occurs at 494 nm and emission at 521nm.
Fluorescein has a pKa of 6.4 and multiple ionization equilibria. This leads to pH dependent absorption and emission over the range of 5 to 9. Also, the fluorescence lifetimes of the protonated and deprotonated forms of fluorescein are approximately 3 and 4 ns, which allows for pH determination from non-intensity based measurements. The lifetimes can be recovered using time-correlated single photon counting or phase-modulation fluorimetry.
2. Derivatives
There are many fluorescein derivatives, for example fluorescein isothiocyanate, often abbreviated as FITC. FITC is the original fluorescein molecule functionalized with an isothiocyanate group (-N=C=S), replacing a hydrogen atom on the bottom ring of the structure. This derivative is reactive towards amine groups on proteins inside cells. A succinimidyl-ester functional group attached to the fluorescein core, creating NHS-fluorescein, forms another common amine reactive derivative.
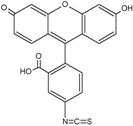
Fluorescein isothiocyanate
Other derivatives of fluorescein include Oregon Green, Tokyo Green, SNAFL, and carboxynaphthofluorescein. These derivatives, along with newer fluors such as Alexa 488 and DyLight 488, have been tailored for various chemical and biological applications where higher photostability, different spectral characteristics, or different attachment groups are needed.
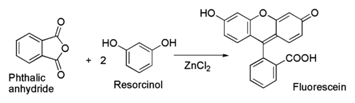
3. Synthesis
Fluorescein can be prepared from phthalic anhydride and resorcinol in the presence of zinc chloride via the Friedel-Crafts reaction.
A second method to prepare fluorescein uses methanesulfonic acid as the catalyst.
4. Application in biological research
In cellular biology, the isothiocyanate derivative of fluorescein is often used to label and track cells in fluorescence microscopy applications (for example, flow cytometry). Additional biologically active molecules (such as antibodies) may also be attached to fluorescein, allowing biologists to target the fluorophore to specific proteins or structures within cells. This application is common in yeast display.
Fluorescein can also be conjugated to nucleoside triphosphates and incorporated into a probe for in situ hybridisation. Fluorescein-labelled probes can be imaged using FISH, or targeted by antibodies using immunohistochemistry. The latter is a common alternative to digoxigenin, and the two are used together for labelling two genes in one sample.
Fluorescent labelling
Fluorescent labeling is the process of covalently attaching a fluorophore to another molecule, such as a protein or nucleic acid. This is generally accomplished using a reactive derivative of the fluorophore that selectively binds to a functional group contained in the target molecule. The most commonly labelled molecules are antibodies, which are then used as specific probes for detection of a particular target.
1. Detection
Fluorescent labels are generally used for detection of a protein or other labelled molecule via a fluorescence microscope, flow cytometer or some other fluorescence reading instrument. These can be useful in localization of a target within a cell, flow cytometry (FACS) analysis, western blot assays, and other immunoanalytical methods.
2. Labelling techniques
Fluorescent labelling is accomplished using a chemically reactive derivative of a fluorophore. Common reactive groups include amine reactive isothiocyanate derivatives such as FITC and TRITC (derivatives of fluorescein and rhodamine), amine reactive succinimidyl esters such as NHS-fluorescein, and sulfhydryl reactive maleimide activated fluors such as fluorescein-5-maleimide. Reaction of any of these reactive dyes with another molecule results in a stable covalent bond formed between a fluorophore and a labelled molecule.
Reactive fluorescent dyes are available from many sources (see below). They can be obtained with diffferent reactive groups for attachment to various functional groups within the target molecule. They are also available in labelling kits that contain all the components to carry out a labelling reaction.
Acetylation
Acetylation (or in IUPAC nomenclature ethanoylation) describes a reaction that introduces an acetyl functional group into an organic compound. Deacetylation is the removal of the acetyl group.
Moreover, it is that process of introducing an acetyl group into a compound, specifically, the substitution of an acetyl group for an active hydrogen atom. A reaction involving the replacement of the hydrogen atom of a hydroxyl group with an acetyl group (CH3 CO) yields a specific ester, the acetate. Acetic anhydride is commonly used as an acetylating agent reacting with free hydroxyl groups.
Acetylation of proteins
In biology, i.e. in living cells, acetylation occurs as a co-tranlational and post-translational modification of proteins, for example, histones and tubulins.
1. N-alpha-terminal Acetylation
Acetylation of the N-terminal alpha-amine of proteins is a whidespread modification in eucaryotes. 40-50% of yeast proteins, and 80-90% of human proteins are modified in this manner, and the pattern of modification is found to be conserved throughout evolution. The modification is performed by N-alpha-acetyltransferases (NATs), a sub-family of the GNAT superfamily of acetyltransferases, which also include histone acetyl transferases. The GNATs transfer the acetylgroup from acetyl-coenzyme A to the amine group. The NATs have been most extensively studied in yeast. Here three NAT complexes, NatA/B/C, have been found to perform most N-alpha-terminal acetylations. They have sequence specificity for their substrates, and it is believed that they are associated with the ribosome, where they acetylate nascent polypeptides co-translationally.
In humans, only one NAT complex , the human NatA, has been identified and characterized. Subunits of the human NatA complex have been coupled to cancer-related processes such as hypoxia-response and the beta-catenin pathway. It has been found to be over-expressed in papillary thyroid carcinoma and neuroblastoma.
Despite being such a conserved and whidespread modification, little is known about the biological role of N-alpha-terminal acetylation. Proteins such as actin and tropomyosin has been found to be dependent of NatB acetylation to form proper actin filaments. This is yet only an example of the potential importance of this modification.
2. Histone Acetylation and Deacetylation
In histone acetylation and deacetylation, the histones are acetylated and deacetylated on lysine residues in the N-terminal tail as part of gene regulation.
Typically, these reactions are catalyzed by enzymes with “histone acetyltransferase” (HAt) or “histone deacetylase” (HDAc) activity.
3. Tubulin Acetylation and Deacetylation
Tubulin Acetylation and Deacetylation system is well worked out in Chlamydomonas. A Tubulin acetyltransferase located in the axoneme acetylates a specific lysine residue in the α-tubulin subunit in assembled microtubule. Once disassembled, this acetylation can be removed by another specific deacetylase which is cytosolic. Thus the axonemal microtubules (long half life) carry this signature acetylation absent from cytosolic microtubules (short half life).
Amidation
In chemistry, an amide is one of two kinds of compounds:
• the organic functional group characterized by a carbonyl group (C=O) linked to a nitrogen atom (N), or a compound that contains this functional group (pictured to the right); or
• a particular kind of nitrogen anion.
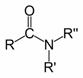
Amide functional group
Amides are the most stable of all the carbonyl functional groups.
Many chemists make a pronunciation distinction between the two, saying (IPA: [ə’mɪd] for the carbonyl-nitrogen compound and [‘æmɑɪd] for the anion. Others substitute one of these pronunciations with [‘æmɪd], while still others pronounce both as [‘æmɪd], making them homonyms.
In the first sense referred to above, an amide is an amine where one of the nitrogen substituents is an acyl group; it is generally represented by the formula: R1(CO)NR2R3 , where either or both R2 and R3 may be hydrogen. Specifically, an amide can also be regarded as a derivative of a carboxylic acid in which the hydroxyl group has been replaced by an amine or ammonia.
Compounds in which a hydrogen atom on nitrogen from ammonia or an amine is replaced by a metal cation are also known as amides or azanides.
The second sense of the word amide is the amide anion, which is a deprotonated form of ammonia (NH3) or an amine. It is generally represented by the formula: [R1NR2]-, and is an extremely strong base, due to the extreme weakness of ammonia and its analogues as Brønsted acids.
The remainder of this article is about the carbonyl-nitrogen sense of amide. For examples of the anionic amide, see the articles Sodium amide and Lithium diisopropylamide.
Contents
1. Amide synthesis
2. Amide reactions
3. Amide linkage (peptide bond)
4. Amide properties
5. Solubility
6. Derivatives
7. Naming conventions
8. References
9. External links
1. Amide synthesis
• Amides are commonly formed from the reaction of a carboxylic acid with an amine. This is the reaction that forms peptide bonds between amino acids. These amides can participate in hydrogen bonding as hydrogen bond acceptors and donors, but do not ionize in aqueous solution, whereas their parent acids and amines are almost completely ionized in solution at neutral pH. Amide formation plays a role in the synthesis of some condensation polymers, such as nylon and Aramid (Twaron / Kevlar). In biochemistry peptides are synthesized in solid phase peptide synthesis.

• Cyclic amides are synthesized in the Beckmann rearrangement from oximes.
• Amides also form ketones in the Schmidt reaction
• Amides can be prepared from aryl alkyl ketones, sulfur and morpholine in the Willgerodt-Kindler reaction
• Other amide-forming reactions are the Passerini reaction and the Ugi reaction
• In the Bodroux reaction an amide RNHCOR’ is synthesized from a carboxylic acid R-COOH and the the adduct of a Grignard reagent with an aniline derivative ArNHR’ [1]
• In the Chapman rearrangement (first reported in 1925) an aryl imino ester is converted to a N,N-diaryl amide:
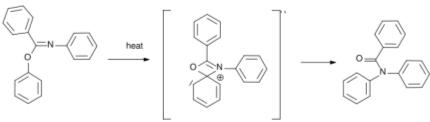
The reaction mechanism is based on a nucleophilic aromatic substitution. [2]
2. Amide reactions
• Amide breakdown is possible via amide hydrolysis. Such hydrolysis can occur under basic or acidic conditions. Acidic conditions yield the carboxylic acid and the ammonium ion while basic hydrolysis yield the carboxylate ion and ammonia.
• In the Vilsmeier-Haack reaction an amide is converted into an imine.
• Hofmann rearrangement of primary amides to primary amines.
Owing to their resonance stabilization, amides are relatively unreactive under physiological conditions, even less than similar compounds such as esters. Nevertheless, amides can undergo chemical reactions, usually through an attack of an electronegative atom on the carbonyl carbon, breaking the carbonyl double bond and forming a tetrahedral intermediate. When the functional group attacking the amide is a thiol, hydroxyl or amine, the resulting molecule may be called a cyclol or, more specifically, a thiacyclol, an oxacyclol or an azacyclol, respectively.
The proton of an amide does not dissociate readily under normal conditions; its pKa is usually well above 15. However, under extremely acidic conditions, the carbonyl oxygen can become protonated with a pKa of roughly -1.
Amides will react with nitrous acid (HONO) forming the carboxylic acid and yielding nitrogen. Nitrous acid is formed by addition of a strong acid to a nitrate (III) salt in solution at temperatures of between 0 and 10 degrees.
Amides undergo Hofmann’s degradation reaction in which an amide yields an amine with one less carbon atom upon reaction with bromine and sodium hydroxide. One should also note that reacting the amide with the strong reducing agent lithium tetrahidridoaluminate yields an amine with the same number of carbon atoms.
Amides are dehydrated with phosphorus (V) oxide forming the nitrile. Care should be taken when perfoming such a reaction since phosphorus (V) oxide smoulders when in contact with organic matter.
3. Amide linkage (peptide bond)
An amide linkage is kinetically stable to hydrolysis. However, it can be hydrolysed in boiling alkali, as well as in strong acidic conditions. Amide linkages in a biochemical context are called peptide linkages. Amide linkages constitute a defining molecular feature of proteins, the secondary structure of which is due in part to the hydrogen bonding abilities of amides.
4. Amide properties
Compared to amines, amides are very weak bases. While the conjugate acid of an amine has a pKa of about 9.5, the conjugate acid of an amide has a pKa around -0.5. Therefore amides don’t have as clearly noticeable acid-base properties in water. This lack of basicity is explained by the electron-withdrawing nature of the carbonyl group where the lone pair of electrons on the nitrogen is delocalized by resonance, thus forming a partial double bond with the carbonyl carbon and putting a negative charge on the oxygen. On the other hand, amides are much stronger bases than carboxylic acids, esters, aldehydes, and ketones (conjugated acid pKa between -6 and -10). It is estimated in silico that acetamide is represented by resonance structure A for 62% and by B for 28% [3]. Resonance is largely prevented in the very strained quinuclidone.

5. Solubility
Amides contain carbonyl (C=O) and ether (N-C) dipoles arising from covalent bonding between electronegative oxygen and nitrogen atoms and electro-neutral carbon atoms. Primary and secondary amides also contain two- and one N-H dipoles, respectively. Because of the pi-bonding arrangement of the carbonyl and the greater electronegativity of oxygen, the carbonyl (C=O) is a stronger dipole than the N-C dipole. The presence of a C=O dipole and, to a lesser extent a N-C dipole, allows amides to act as H-bond acceptors. In primary and secondary amides, the presence of N-H dipoles allows amides to function as H-bond donors as well. Thus amides can participate in hydrogen bonding with water and other protic solvents; the oxygen and nitrogen atoms can accept hydrogen bonds from water and the N-H hydrogen atoms can donate H-bonds. As a result of interactions such as these, the water solubility of amides is greater than that of corresponding hydrocarbons
While hydrogen bonding may enhance the water solubility of amides relative to hydrocarbons (alkanes, alkenes, alkynes and aromatic compounds), amides typically are regarded as compounds with low water solubility. They are significantly less water soluble than comparable acids or alcohols due to: 1). their non-ionic character 2). the presence of nonpolar hydrocarbon functionality, and 3). the inability of tertiary amides to donate hydrogen bonds to water (they can only be H-bond acceptors). Thus amides have water solubilities roughly comparable to esters. Typically amides are less soluble than comparable amines and carboxylic acids since these compounds can both donate and accept hydrogen bonds, and can ionize at appropriate pHs to further enhance solubility
6. Derivatives
Sulfonamides are analogues of amides in which the atom double-bonded to oxygen is sulfur rather than carbon.
Cyclic amides are called lactams.
7. Naming conventions
• Example: CH3CONH2 is named acetamide or ethanamide
• Other examples: propan-1-amide, N,N-dimethylpropanamide, acrylamide
• For more detail see IUPAC nomenclature of organic chemistry – Amines and Amides
8. References
1. Bodroux F., Bull. Soc. Chim. France, 1905, 33, 831;
2. Advanced organic Chemistry, Reactions, mechanisms and structure 3ed. Jerry March ISBN 0-471-85472-7
3. “Amide Resonance” Correlates with a Breadth of C-N Rotation Barriers Carl R. Kemnitz and Mark J. Loewen J. Am. Chem. Soc.; 2007; 129(9) pp 2521 – 2528.
Palmitoylation
Palmitoylation is the covalent attachment of fatty acids, such as palmitic acid, to cysteine residues of membrane proteins.[1]
The precise function of palmitoylation depends on the particular protein being considered. Palmitoylation enhances the hydro-phobicity of proteins and contributes to their membrane association. Palmitoylation also appears to play a significant role in subcellular trafficking of proteins between membrane compartments, as well as in modulating protein-protein interactions.[2]
References
1. Linder, M.E., “Reversible modification of proteins with thioester-linked fatty acids,” Protein Lipidation, F. Tamanoi and D.S. Sigman, eds., pp. 215-40 (San Diego, CA: Academic Press, 2000).
2. Basu, J., “Protein palmitoylation and dynamic modulation of protein function,” Current Science, Vol. 87, No. 2, pp. 212-17 (25 July 2004).
Myristoylation
Myristoylation is an irreversible, post-translational protein modification found in animals, plants, fungi and viruses. In this protein modification a myristoyl group (derived from myristic acid) is covalently attached via an amide bond to the alpha-amino group of an N-terminal glycine residue of a nascent polypeptide. The modification is catalyzed by the enzyme N-myristoyltransferase, and occurs most commonly on glycine residues exposed during co-translational N-terminal methionine removal. Myristoylation also occurs post-translationally, for example when previously internal glycine residues become exposed by caspase cleavage during apoptosis.
1. Function
Myristoylation plays a vital role in membrane targeting and signal transduction in plant responses to environmental stress.
2. References
• Podell S and Gribskov M. (2004) “Predicting N-terminal myristoylation sites in plant proteins”, BMC Genomics, 5, 37.
• Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ (2000) “Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis”, Science 290, 1761-1765.
Phosphorylation
Phosphorylation is the addition of a phosphate (PO4) group to a protein molecule or a small molecule. Another way to define it would be the introduction of a phosphate group into an organic molecule. Its prominent role in biochemistry is the subject of a very large body of research (as of January 2006, the Medline database returns over 120,000 articles on the subject, largely on protein phosphorylation).
Contents
1. Protein phosphorylation
1.1. Function
1.2. Signaling networks
1.3. Protein phosphorylation sites
1.4. Types of phosphorylation
1.5. Detection and characterization
1.6. History
2. Other kinds
3. External links
1. Protein phosphorylation
1.1. Function
In eukaryotes, protein phosphorylation is probably the most important regulatory event. Many enzymes and receptors are switched “on” or “off” by phosphorylation and dephosphorylation. Phosphorylation is catalyzed by various specific protein kinases, whereas phosphatases dephosphorylate.
Adding a phosphoryl (PO3) to a polar R group of an amino acid might not seem like it would do much to a protein, but it can actually turn a nonpolar hydrophobic protein into a polar and extremely hydrophilic molecule. Phosphorylation is observed on serine, threonine, tyrosine and histidine residues.
An example of the important role that phosphorylation plays is the p53 tumor suppressor gene, which—when active—stimulates transcription of genes that suppress the cell cycle, even to the extent that it undergoes apoptosis. However, this activity should be limited to situations where the cell is damaged or physiology is disturbed. To this end, the p53 protein is extensively regulated. In fact, p53 contains more than 18 different phosphorylation sites.
Upon the deactivating signal, the protein becomes dephosphorylated again and stops working. This is the mechanism in many forms of signal transduction, for example the way in which incoming light is processed in the light-sensitive cells of the retina.
1.2. Signaling networks
The network underlying phosphorylation can be very complex. In some cellular signalling pathways, a protein A phosphorylates B, and B phosphorylates C, but A also phosphorylates C directly, and B can phosphorylate D, which may in turn phosphorylate A. Global approaches to identify and quantify phosphorylated proteins, like mass spectrometry-based proteomics, are becoming increasingly important for the systematic analysis of complex phosphorylation networks. For example, one study has identified dynamic changes in the phosphorylation status of more than 6000 sites after stimulation with epidermal growth factor. Analysis of phosphoproteins is a branch of proteomics called phosphoproteomics.
1.3. Protein phosphorylation sites
There are thousands of distinct phosphorylation sites in a given cell since: 1) There are thousands of different kinds of proteins in any particular cell (such as a lymphocyte). 2) It is estimated that 1/10th to 1/2 of proteins are phosphorylated (in some cellular state). 3) Phosphorylation often occurs on multiple distinct sites on a given protein.
Since phosphorylation of any site on a given protein can change the function or localization of that protein, understanding the “state” of a cell requires knowing the phosphorylation state of its proteins. For example, if amino acid Serine-473 (“S473”) in the protein AKT is phosphorylated AKT is generally functionally active as a kinase. If not, it is an inactive kinase.
1.4. Types of phosphorylation
See also protein kinase for more details on the different types of phosphorylation
Within a protein, phosphorylation can occur on several amino acids. Phosphorylation on serine is the most common, followed by threonine. Tyrosine phosphorylation is relatively rare. However, since tyrosine phosphorylated proteins are relatively easy to purify using antibodies, tyrosine phosphorylation sites are relatively well understood. Histidine and aspartate phosphorylation occurs in prokaryotes as part of two-component signalling.
1.5. Detection and characterization
Antibodies can be used as powerful tools to detect whether a protein is phosphorylated at any particular site. Such antibodies are called phospho-specific antibodies; hundreds of such antibodies are now available. They are becoming critical reagents both for basic research and for clinical diagnosis.
PTM isoforms are easily detected on 2D gels. Indeed, phosphorylation replaces neutral hydroxyl groups on serines, threonines or tyrosines with negatively charged phosphates with pKs near 1.2 and 6.5. Thus, below pH 5.5, phosphates add a single negative charge, near pH 6.5 they add 1.5 negative charges and above pH 7.5 they add 2 negative charges. The relative amount of each isoform can also easily and rapidly be determined from staining intensity on 2D gels.
A detailed characterization of the sites of phosphorylation is very difficult and the quantitation of protein phosphorylation by mass spectrometry requires isotopic internal standard approaches (Gerber et al., 2003). A relative quantitation can be obtained with a variety of differential isotope labeling technologies (Gigy et al., 2002, Goshe et al., 2003).
1.6. History
In 1906, Phoebus A. Levene at the Rockefeller Institute for Medical Research identified phosphate in the protein Vitellin (phosvitin), and by 1933 had detected phosphoserine in Casein, with Fritz Lipmann. However, it took another 20 years before Eugene P. Kennedy described the first ‘enzymatic phosphorylation of proteins’.
2. Other kinds
ATP, the “high-energy” exchange medium in the cell, is synthesized in the mitochondrion by addition of a third phosphate group to ADP in a process referred to as oxidative phosphorylation. ATP is also synthesized by substrate-level phosphorylation during glycolysis. ATP is synthesized at the expense of solar energy by photophosphorylation in the chloroplasts of plant cells.
Phosphorylation of sugars is often the first stage of their catabolism. It allows cells to accumulate sugars because the phosphate group prevents the molecules from diffusing back across their transporter.
Glycosylation
Glycosylation is the process or result of addition of saccharides to proteins and lipids. The process is one of four principal co-translational and post-translational modification steps in the synthesis of membrane and secreted proteins and the majority of proteins synthesized in the rough ER undergo glycosylation. It is an enzyme-directed site-specific process, as opposed to the non-enzymatic chemical reaction of glycation. Two types of glycosylation exist: N-linked glycosylation to the amide nitrogen of asparagine side chains and O-linked glycosylation to the hydroxy oxygen of serine and threonine side chains.
Contents
1. Purpose
2. Mechanisms
2.1. N-linked glycosylation
2.2. O-linked glycosylation
2.2.1. O-N-acetylgalactosamine (O-GalNAc)
2.2.2. O-fucose
2.2.3. O-glucose
2.2.4. O-N-acetylglucosamine (O-GlcNAc)
2.3. GPI anchor
2.4. C-mannosylation
1. Purpose
The polysaccharide chains attached to the target proteins serve various functions. For instance, some proteins do not fold correctly unless they are glycosylated first. Also, polysaccharides linked at the amide nitrogen of asparagine in the protein confer stability on some secreted glycoproteins. Experiments have shown that glycosylation in this case is not a strict requirement for proper folding, but the unglycosylated protein degrades quickly. Glycosylation may play a role in cell-cell adhesion (a mechanism employed by cells of the immune system), as well.
2. Mechanisms
There are various mechanisms for glycosylation, although all share several common features:
Glycosylation is an enzymatic process
The donor molecule is an activated nucleotide sugar
The process is site-specific.
2.1. N-linked glycosylation
N-linked glycosylation is important for the folding of some of eukaryotic proteins. The N-linked glycosylation process occurs in eukaryotes and widely in archaea, but very rarely in bacteria.
For N-linked oligosaccharides, a 14-sugar precursor is first added to the asparagine in the polypeptide chain of the target protein. The structure of this precursor is common to most eukaryotes, and contains 3 glucose, 9 mannose, and 2 N-acetylglucosamine molecules. A complex set of reactions attaches this branched chain to a carrier molecule called dolichol, and then it is transferred to the appropriate point on the polypeptide chain as it is translocated into the ER lumen.
There are two major types of N-linked saccharides: high-mannose oligosaccharides, and complex oligosaccharides (Alberts et al., Ch 13, pg 604). High-mannose is, in essence, just two N-acetylglucosamines with many mannose residues, often almost as many as are seen in the precursor oligosaccharides before it is attached to the protein. Complex oligosaccharides are so named because they can contain almost any number of the other types of saccharides, including more than the original two N-acetylglucosamines. Proteins can be glycosylated by both types of oligos on different portions of the protein. Whether an oligosaccharide is high-mannose or complex is thought to depend on its accessibility to saccharide-modifying proteins in the Golgi. If the saccharide is relatively inaccessible, it will most likely stay in its original high-mannose form. If it is accessible, then it is likely that many of the mannose residues will be cleaved off and the saccharide will be further modified by the addition of other types of group as discussed above.
The oligosaccharide chain is attached by oligosaccharyltransferase to asparagine occurring in the tripeptide sequence Asn-X-Ser or Asn-X-Thr, where X could be any amino acid except Pro. This sequence is known as a glycosylation sequon. After attachment, once the protein is correctly folded, the three glucose residues are removed from the chain and the protein is available for export from the ER. The glycoprotein thus formed is then transported to the Golgi where removal of further mannose residues may take place. However, glycosylation itself does not seem to be as necessary for correct transport targeting of the protein, as one might think. Studies involving drugs that block certain steps in glycosylation, or mutant cells deficient in a glycosylation enzyme, still produce otherwise-structurally-normal proteins that are correctly targeted, and this interference does not seem to interfere severely with the viability of the cells. Mature glycoproteins may contain a variety of oligomannose N-linked oligosaccharides containing between 5 and 9 mannose residues. Further removal of mannose residues leads to a ‘core’ structure containing 3 mannose, and 2 N-acetylglucosamine residues, which may then be elongated with a variety of different monosaccharides including galactose, N-acetylglucosamine, N-acetylgalactosamine, fucose and sialic acid.
2.2. O-linked glycosylation
2.2.1. O-N-acetylgalactosamine (O-GalNAc)
O-linked glycosylation occurs at a later stage during protein processing, probably in the Golgi apparatus. This is the addition of N-acetyl-galactosamine to serine or threonine residues by the enzyme UDP-N-acetyl-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase, followed by other carbohydrates (such as galactose and sialic acid). This process is important for certain types of proteins such as proteoglycans, which involves the addition of glycosaminoglycan chains to an initially unglycosylated “proteoglycan core protein.” These additions are usually serine O-linked glycoproteins, which seem to have one of two main functions. One function involves secretion to form components of the extracellular matrix, adhering one cell to another by interactions between the large sugar complexes of proteoglycans. The other main function is to act as a component of mucosal secretions, and it is the high concentration of carbohydrates that tends to give mucus its “slimy” feel. Proteins that circulate in the blood are not normally O-glycosylated, with the exception of IgA1 and IgD (two types of antibody) and C1-inhibitor.
2.2.2. O-fucose
O-fucose is added between the second and third conserved cysteines of EGF-like repeats in the Notch protein, and possibly other substrates by GDP-fucose protein O-fucosyltransferase 1, and to Thrombospondin repeats by GDP-fucose protein O-fucosyltransferase 2. In the case of EGF-like repeats, the O-fucose may be further elongated to a tetrasaccharide by sequential addition of N-acetylglucosamine (GlcNAc), galactose, and sialic acid, and for Thrombospondin repeats, may be elongated to a disaccharide by the addition of glucose. Both of these fucosyltransferases have been localized to the endoplasmic reticulum, which is unusual for glycosyltransferases, most of which function in the Golgi apparatus.
2.2.3. O-glucose
O-glucose is added between the first and second conserved cysteines of EGF-like repeats in the Notch protein, and possibly other substrates by an unidentified O-glucosyltransferase.
2.2.4. O-N-acetylglucosamine (O-GlcNAc)
O-GlcNAc is added to serines or threonines by O-GlcNAc transferase. O-GlcNAc appears to occur on serines and threonines that would otherwise be phosphorylated by serine/threonine kinases. Thus, if phosphorylation occurs, O-GlcNAc does not, and vice versa. This is an incredibly important finding because phosphorylation/dephosphorylation has become a scientific paradigm for the regulation of signaling within cells. A massive amount of cancer research is focused on phosphorylation. Ignoring the involvement of this form of glycosylation which clearly appears to act in concert with phosphorylation means that a lot of current research is missing at least half of the picture. O-GlcNAc addition and removal also appear to be key regulators of the pathways which are deregulated in diabetes mellitus. The gene encoding the O-GlcNAc removal enzyme has been linked to non-insulin dependent diabetes mellitus. It is the terminal step in a nutrient sensing hexosamine signaling pathway.
2.3. GPI anchor
A special form of glycosylation is the GPI anchor. This form of glycosylation functions to attach a protein to a hydrophobic lipid anchor, via a glycan chain. (see also prenylation)
2.4. C-mannosylation
A mannose sugar is added to tryptophan residues in Thrombospondin repeats. This is an unusual modification both because the sugar is linked to a carbon rather than a reactive atom like a nitrogen or oxygen and because the sugar is linked to a tryptophan residue rather than an asparagine or serine/threonine.
Biotinylation
In biochemistry, biotinylation is the process of covalently attaching a biotin tag to a molecule or surface.
1. Purpose
1.1. Determining extent of biotinylation
Reaction conditions for biotinylation are chosen such that the target molecule (e.g. an antibody) is labelled with enough biotin substituents to purify or detect the molecule, but not so much that the biotin interferes with the function of the molecule. The HABA dye(2-(4-hydroxyazobenzene) benzoic acid) method is used to determine that extent of biotinylation. HABA dye is bound to avidin and yields a characteristic absorbance. When biotin, in the form of biotinylated protein or other molecule, is introduced, it displaces the dye, resulting in a change in absorbance at 500 nm. The absorbance change is directly proportional to the level of biotin in the sample.
1.2. Purification
The biotin tag can be used in affinity chromatography together with a column that has avidin or streptavidin bound to it, which is the natural chelator for biotin.
1.3. Detection
This tag can also be used in detection of the protein via anti-biotin antibodies or avidine/streptavidin tagged detectors like horseradish peroxidase or a fluorescent dye. This can be useful in localization, ELISA assays, ELISPOT assays, western blots and other immunoanalytical methods.
2. Other uses
The non-covalent bond formed between biotin and avidin or streptavidin has a binding affinity that is tighter than most antigen and antibody bonds and approaches the strength of a covalent bond. This very tight binding makes labeling proteins with biotin a useful tool for applications such as affinity chromatography using immobilized avidin or streptavidin to separate the biotinylated protein from a mixture of other proteins and biochemicals. Biotinylated protein such as biotinylated bovine serum albumin (BSA) is used in solid-phase assays as a coating on the well surface in multiwell assay plates. Biotinylation of red blood cells has been used as a means of determining total blood volume without the use of radiolabels such as chromium 51, allowing volume determinations in low birth weight infants and pregnant women who could not otherwise be exposed to the required doses of radioactivity.
Farnesylation
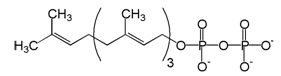
A post-translational modification of proteins by the attachment of an isoprenoid to the C-terminal cysteine residue. The isoprenoids used, farnesyl diphosphate or geranylgeranyl diphosphate, are derived from the same biochemical pathway that produces cholesterol.
1. Farnesyl pyrophosphate
Farnesyl pyrophosphate (FPP) is an intermediate in the HMG-CoA reductase pathway used by organisms in the biosynthesis of terpenes and terpenoids.
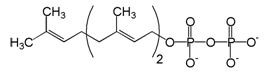
It is the immediate precursor of squalene (via the enzyme squalene synthase), dehydrodolichol diphosphate (a precursor of dolichol), and geranylgeranyl pyrophoshate (GGPP).
1.1. Biosynthesis
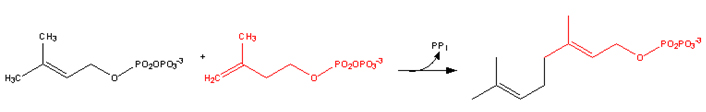
Farnesyl pyrophosphate synthase (a prenyl transferase) catalyzes sequential condensation reactions of dimethylallyl pyrophosphate with 2 units of 3-isopentenyl pyrophosphate to form farnesyl pyrophosphate.
• Dimethylallyl pyrophosphate reacts with 3-isopentenyl pyrophosphate to form geranyl pyrophosphate:
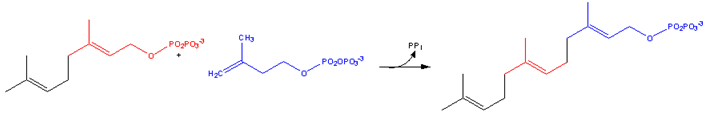
2. Geranylgeranyl pyrophosphate
Geranylgeranyl pyrophosphate is an intermediate in the HMG-CoA reductase pathway used by organisms in the biosynthesis of terpenes and terpenoids. In plants it is additionally the precursor to carotenoids, gibberellins, tocopherols, and chlorophylls.